각각의 기능은 모듈화되어 쉽게 추가하고 관리할 수 있습니다.
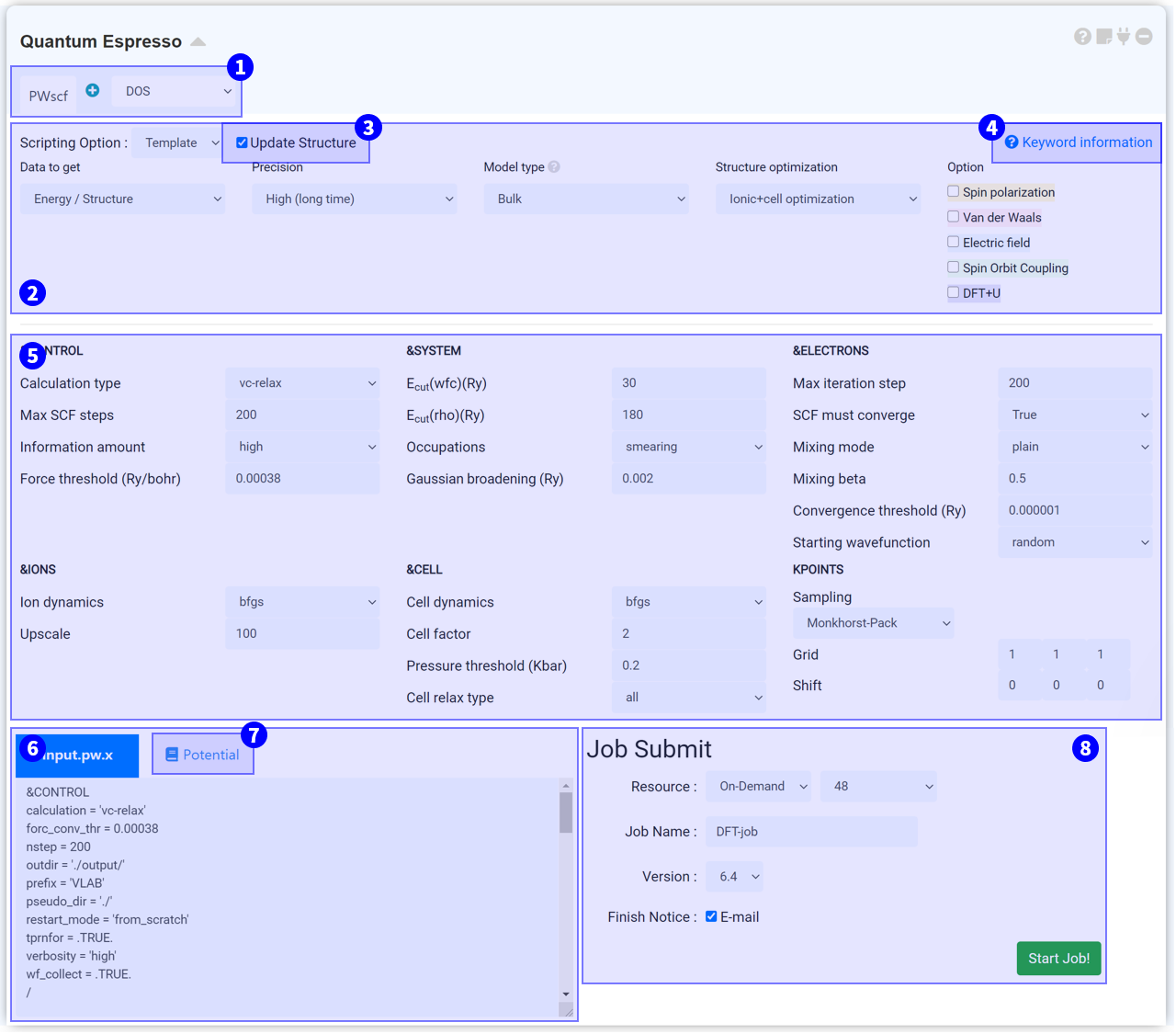
시뮬레이션 모듈인 Quantum Espresso (QE) 모듈은 오픈소스 DFT 계산 소프트웨어 패키지인 Quantum Espresso를 이용하여 전자 구조 계산 (electronic structure calculation)을 수행하기 위한 모듈입니다. MatSQ는 복잡한 코드 공부 없이 클릭 몇 번만으로 DFT 계산을 쉽게 수행할 수 있도록 직관적인 그래픽 인터페이스 (GUI)를 제공하고 있습니다. 대부분의 계산은 설정된 기본 변수에서 cutoff energy, k-point와 같은 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 이외에 숙련된 사용자를 위해 script를 직접 수정할 수 있는 manual 모드도 제공하고 있습니다.
Quantum Espresso에 대한 자세한 정보는 appendix 혹은 www.quantum-espresso.org 에서 확인할 수 있습니다.
Solver란 Quantum Espresso에서 사용하는 계산 패키지를 말합니다. 각 solver는 특정한 역할을 하며, scf 계산을 위한 PWscf (pw.x)와 post-processing solver로 나눌 수 있습니다. PWscf는 모든 데이터를 얻을 때 가장 기본이 되는 solver로, DFT 기반으로 Plane-Wave (PW) basis set 및 pseudopotentials (PP, psp)를 사용하여 전자 구조 특성에 대해 self-consistent 계산을 수행합니다. 어떠한 후처리 데이터를 얻고자 할 때 PWscf 계산을 필수적으로 수행해야 하며, 따라서 QE 모듈은 PWscf를 중심으로 구성되어 있습니다.
post-processing 계산을 수행하는 방법은 다음과 같습니다.
이때 이미 PWscf 계산이 종료된 QE 모듈에서도 post-processing solver를 추가하고 Start Job! 버튼을 누르면 post-processing data를 추가적으로 얻을 수 있습니다.
각 solver의 input script 설정에 대한 정보는 appendix 혹은 ④ Keyword Information 에서 확인할 수 있습니다.
Scripting option을 누르면 Template, General, Manual 옵션을 볼 수 있습니다. Template은 Quantum espresso의 키워드 설정에 익숙하지 않은 분들에게 적합한 옵션입니다. Template에서는 세 가지 질문에 답하여 input script를 설정합니다.
Quantum espresso 모듈은 연결된 structure builder에서 구조 정보를 가져오며, structure builder의 구조 정보가 바뀌면 QE 모듈에도 바뀐 구조 정보가 업데이트됩니다. QE 모듈의 구조정보를 더 이상 업데이트하고 싶지 않은 경우 이 체크박스를 해제하십시오.
각각의 solver는 서로 다른 input parameter를 갖습니다. 자세한 정보를 보기 위해 ? Keyword information 을 클릭하면 다음과 같은 url로 연결됩니다.
MatSQ에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있습니다. Quantum espresso 모듈에 기본적으로 설정된 input parameter는 일반적으로 적절한 값을 설정해 둔 값으로, 대부분의 계산은 cutoff energy, k-point와 같은 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 그러나 논문 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input parameter에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다.
자세한 정보는 appendix 혹은 ④ Keyword Information를 참고하십시오.
QE 모듈을 structure builder에 연결하고 Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ⑤ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다. Input script 창에서 텍스트를 수정하면 자동으로 Manual 모드로 변경됩니다.
Quantum espresso를 이용해 DFT 계산을 수행할 때 해당 원소의 거동을 결정하는 것이 바로 pseudopotential입니다. Pseudopotential 탭에서 돋보기 버튼을 누르면 사용할 수 있는 pseudopotential 리스트를 볼 수 있습니다.
Pseudopotential에 대한 자세한 정보는 Quantum Espresso official documentation 을 참고하십시오.
①~⑦의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
계산이 종료된 QE 모듈에 새로운 QE 모듈을 연결하면, 계산이 종료된 지점에서 계산을 이어 시작하는 restart 계산을 진행할 수 있습니다. 특히 사용자가 임의로 중단한 계산을 되살리고 싶을 때 restart를 통해 계산을 이어 시작할 수 있습니다. 단, 오류가 발생한 계산으로부터는 재시작할 수 없습니다. 다음 경우에 적용하면 좋습니다.
'1. This job is not converged 메시지가 표시됐을 때' 는 iteration (전자 구조 최적화) 중 수렴에 실패하는 것이므로 max electron steps를 늘려 계산하는 것이 좋습니다.
'2. This job is normally finished 메시지를 확인했지만 scf step이 설정한 max scf step에 도달했을 때' 는 iteration 중에는 수렴했지만 설정해준 scf 단계 (원자 위치 최적화)에서 최대 max steps에 도달할 때까지 수렴이 완료되지 못했을 확률이 높으므로 max steps를 늘려 계산하는 것이 좋습니다. 그러나 normally finished 메시지를 볼 때까지 restart하는 것은 좋지 못합니다. restart를 수행하여 수백 step이 진행되는 동안 수렴을 달성하지 못했다면 초기 구조를 변경하여 새로운 계산을 시작하거나, 더 낮은 정확도를 갖는 input parameter set으로 새 계산을 시작하여 단계적으로 정확도를 높이며 계산하는 것이 좋습니다.
3, 4는 완료된 PWscf (pw.x) 계산으로부터 k-point를 목적에 맞게 재배치하여 계산하기 위해 restart를 수행하는 것입니다. 3은 calculation type을 nscf로 변경하고, k-point grid를 보다 큰 값으로 설정합니다. nscf 계산은 non-self consistent 계산을 의미하며, k-point 밀도만 변화시키는 것이기 때문에 같은 k-point 밀도일 때 scf (relax) 계산보다 계산 자원을 절약할 수 있습니다.
4는 calculation type을 bands로 바꾸고, k-point를 원하는 대로 변경하십시오. 이때 k-point 옵션을 crystal(_b)로 선택하여 high-symmetric point를 샘플링하는 것이 좋습니다.
단, 이때 GAMMA 옵션으로 계산된 계산을 'k-point option: {automatic}'으로 restart할 수는 없으니 주의해야 합니다.
Quantum Espresso에 대한 자세한 정보는 appendix 혹은 www.quantum-espresso.org 에서 확인할 수 있습니다.
Solver란 Quantum Espresso에서 사용하는 계산 패키지를 말합니다. 각 solver는 특정한 역할을 하며, scf 계산을 위한 PWscf (pw.x)와 post-processing solver로 나눌 수 있습니다. PWscf는 모든 데이터를 얻을 때 가장 기본이 되는 solver로, DFT 기반으로 Plane-Wave (PW) basis set 및 pseudopotentials (PP, psp)를 사용하여 전자 구조 특성에 대해 self-consistent 계산을 수행합니다. 어떠한 후처리 데이터를 얻고자 할 때 PWscf 계산을 필수적으로 수행해야 하며, 따라서 QE 모듈은 PWscf를 중심으로 구성되어 있습니다.
post-processing 계산을 수행하는 방법은 다음과 같습니다.
- QE 모듈을 추가하고, ① Solver에서 원하는 데이터를 얻는 데 적합한 solver (DOS, charge density, ...)를 클릭하십시오.
- 다음으로 + 버튼을 눌러 PWscf 계산 옆에 새 solver 탭을 추가합니다. Post-processing solver는 원하는 만큼 추가할 수 있습니다.
- Start Job! 버튼을 누르면 PWscf 계산과 함께 추가된 모든 post-processing 작업들이 실행됩니다.
이때 이미 PWscf 계산이 종료된 QE 모듈에서도 post-processing solver를 추가하고 Start Job! 버튼을 누르면 post-processing data를 추가적으로 얻을 수 있습니다.
각 solver의 input script 설정에 대한 정보는 appendix 혹은 ④ Keyword Information 에서 확인할 수 있습니다.
Scripting option을 누르면 Template, General, Manual 옵션을 볼 수 있습니다. Template은 Quantum espresso의 키워드 설정에 익숙하지 않은 분들에게 적합한 옵션입니다. Template에서는 세 가지 질문에 답하여 input script를 설정합니다.
- Target property: 어떠한 데이터를 얻고 싶은지 선택하십시오. 선택에 맞추어 적절한 solver가 자동으로 추가됩니다.
- Precision: 계산의 정확도를 설정합니다.
- Model type: 이전 질문에서 High precision을 선택하면 이 질문이 표시됩니다. 사용할 simulation model이 어떤 분류에 속하는지 선택해 주십시오. k-point grid 설정이 달라집니다.
- Structure optimization: Calculation type을 정하는 질문입니다.
- Fully optimized : calculation type이 scf로 설정되며, 계산 과정 중 초기 구조를 변경하지 않습니다 (static calculation).
- Need optimization : 원자 위치를 조금씩 변경하며 potential surface에서 local minimum을 찾으며 원자 위치 최적화를 진행합니다.
- Need ionic+cell optimization : 원자 위치와 셀 크기를 조금씩 변경하며 potential surface에서 local minimum을 찾으며 원자 위치 및 부피 최적화를 진행합니다.
- Option: 계산에 어떠한 추가 옵션을 고려할 것인지 결정합니다.
Quantum espresso 모듈은 연결된 structure builder에서 구조 정보를 가져오며, structure builder의 구조 정보가 바뀌면 QE 모듈에도 바뀐 구조 정보가 업데이트됩니다. QE 모듈의 구조정보를 더 이상 업데이트하고 싶지 않은 경우 이 체크박스를 해제하십시오.
각각의 solver는 서로 다른 input parameter를 갖습니다. 자세한 정보를 보기 위해 ? Keyword information 을 클릭하면 다음과 같은 url로 연결됩니다.
- PWscf (pw.x): https://www.quantum-espresso.org/Doc/INPUT_PW.html
- DOS (projwfc.x): https://www.quantum-espresso.org/Doc/INPUT_PROJWFC.html
- Charge Denstiy (pp.x): https://www.quantum-espresso.org/Doc/INPUT_PP.html
- Band Structure (bands.x): https://www.quantum-espresso.org/Doc/INPUT_BANDS.html
MatSQ에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있습니다. Quantum espresso 모듈에 기본적으로 설정된 input parameter는 일반적으로 적절한 값을 설정해 둔 값으로, 대부분의 계산은 cutoff energy, k-point와 같은 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 그러나 논문 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input parameter에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다.
자세한 정보는 appendix 혹은 ④ Keyword Information를 참고하십시오.
QE 모듈을 structure builder에 연결하고 Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ⑤ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다. Input script 창에서 텍스트를 수정하면 자동으로 Manual 모드로 변경됩니다.
Quantum espresso를 이용해 DFT 계산을 수행할 때 해당 원소의 거동을 결정하는 것이 바로 pseudopotential입니다. Pseudopotential 탭에서 돋보기 버튼을 누르면 사용할 수 있는 pseudopotential 리스트를 볼 수 있습니다.
Pseudopotential에 대한 자세한 정보는 Quantum Espresso official documentation 을 참고하십시오.
①~⑦의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
계산이 종료된 QE 모듈에 새로운 QE 모듈을 연결하면, 계산이 종료된 지점에서 계산을 이어 시작하는 restart 계산을 진행할 수 있습니다. 특히 사용자가 임의로 중단한 계산을 되살리고 싶을 때 restart를 통해 계산을 이어 시작할 수 있습니다. 단, 오류가 발생한 계산으로부터는 재시작할 수 없습니다. 다음 경우에 적용하면 좋습니다.
- This job is not converged 메시지가 표시됐을 때
- This job is normally finished 메시지를 확인했지만 scf step이 설정한 max scf step에 도달했을 때 ((vc-)relax의 경우)
- DOS 계산을 위해 k-point를 늘려 계산하고 싶을 때 (nscf)
- Band structure 계산을 위해 k-point를 high-symmetric point 경로로 설정하고 싶을 때 (bands)
'1. This job is not converged 메시지가 표시됐을 때' 는 iteration (전자 구조 최적화) 중 수렴에 실패하는 것이므로 max electron steps를 늘려 계산하는 것이 좋습니다.
'2. This job is normally finished 메시지를 확인했지만 scf step이 설정한 max scf step에 도달했을 때' 는 iteration 중에는 수렴했지만 설정해준 scf 단계 (원자 위치 최적화)에서 최대 max steps에 도달할 때까지 수렴이 완료되지 못했을 확률이 높으므로 max steps를 늘려 계산하는 것이 좋습니다. 그러나 normally finished 메시지를 볼 때까지 restart하는 것은 좋지 못합니다. restart를 수행하여 수백 step이 진행되는 동안 수렴을 달성하지 못했다면 초기 구조를 변경하여 새로운 계산을 시작하거나, 더 낮은 정확도를 갖는 input parameter set으로 새 계산을 시작하여 단계적으로 정확도를 높이며 계산하는 것이 좋습니다.
3, 4는 완료된 PWscf (pw.x) 계산으로부터 k-point를 목적에 맞게 재배치하여 계산하기 위해 restart를 수행하는 것입니다. 3은 calculation type을 nscf로 변경하고, k-point grid를 보다 큰 값으로 설정합니다. nscf 계산은 non-self consistent 계산을 의미하며, k-point 밀도만 변화시키는 것이기 때문에 같은 k-point 밀도일 때 scf (relax) 계산보다 계산 자원을 절약할 수 있습니다.
4는 calculation type을 bands로 바꾸고, k-point를 원하는 대로 변경하십시오. 이때 k-point 옵션을 crystal(_b)로 선택하여 high-symmetric point를 샘플링하는 것이 좋습니다.
단, 이때 GAMMA 옵션으로 계산된 계산을 'k-point option: {automatic}'으로 restart할 수는 없으니 주의해야 합니다.
Reaction Path (NEB)는 Quantum espresso에서 제공되는 neb.x solver를 이용하여 NEB (Nudged Elastic Band) 계산을 수행하기 위한 모듈입니다.
NEB 계산을 위해 Reaction Path (NEB) 모듈에 연결된 structure builder의 structrue list에 initial, final image (혹은 필요하다면 intermediate image까지)를 추가해 두어야 합니다. 더 나은 결과를 위해 모든 image는 미리 구조 최적화 (structual relaxation/optimization)가 완료된 구조를 사용하는 것이 좋습니다.
NEB Input script 설정에 대한 자세한 정보는 Quantum Espresso Input description (neb.x) 을 참고하십시오.
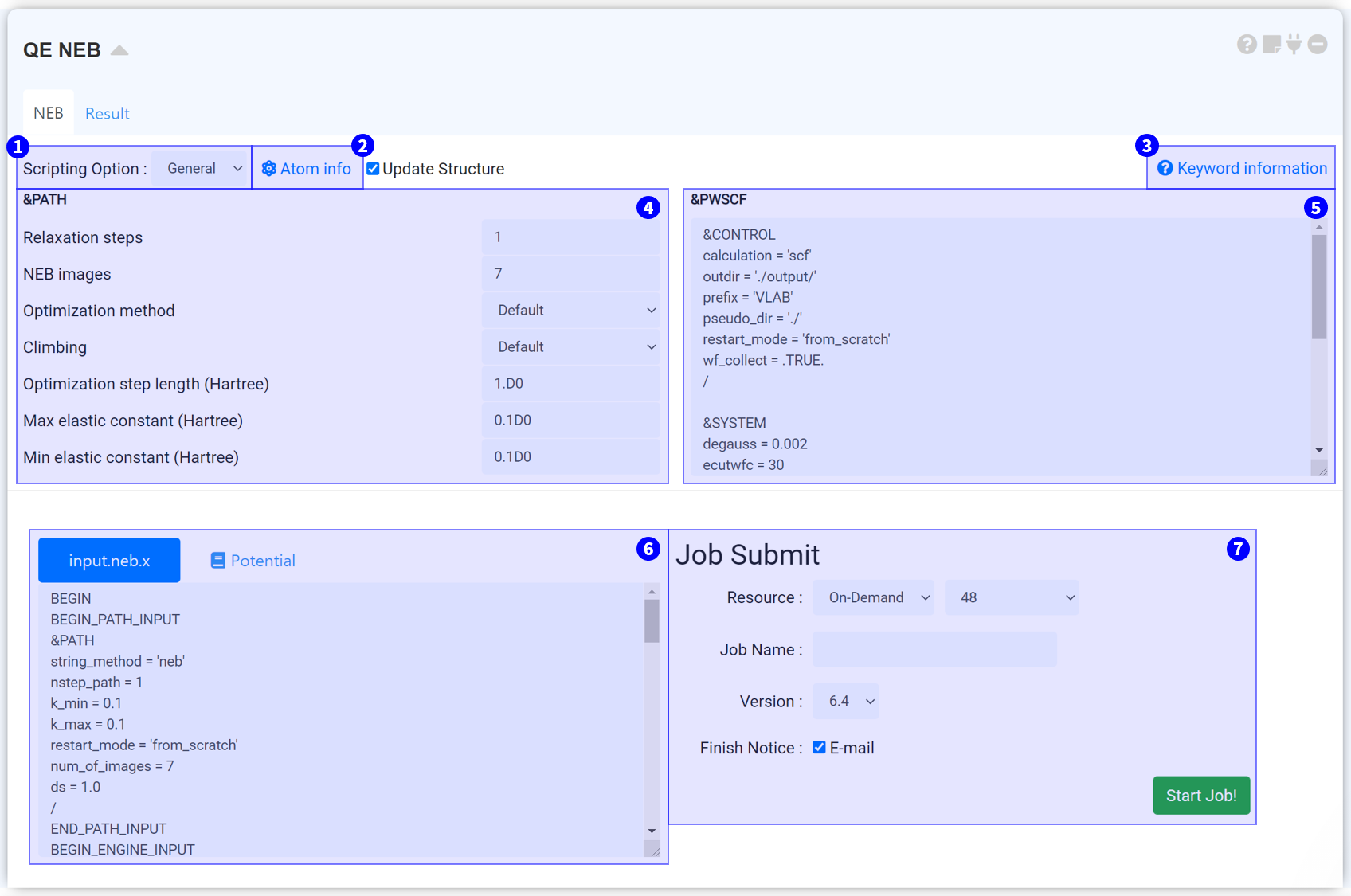
Reaction Path (NEB) 모듈은 General과 Manual 모드를 선택할 수 있습니다.
원자를 고정할 때 쓰는 도구입니다. 연결된 Structure builder의 structure list에 있는 구조의 순서대로 탭이 나뉘어 있습니다. 원하는 원자를 선택하면 해당 원자는 계산 중 고정된 것으로 간주됩니다.
neb.x의 모든 키워드 목록을 볼 수 있는 페이지입니다.
&PATH namelist에 속하는 키워드는 NEB 계산의 조건을 설정하는 키워드입니다. ③ Keyword information을 참고하여 적절하게 설정하십시오. Climbing 키워드를 'auto' 로 설정하면 Climbing NEB 계산을 수행할 수 있습니다.
NEB 경로를 결정할 때 사용되는 키워드입니다. 구조 Relaxation 시 사용한 input script를 복사하여 붙여넣을 수 있습니다.
설정한 input parameter는 input script 형태로 변환되어 서버에 전송됩니다. 잘못된 점은 없는지 확인하십시오.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
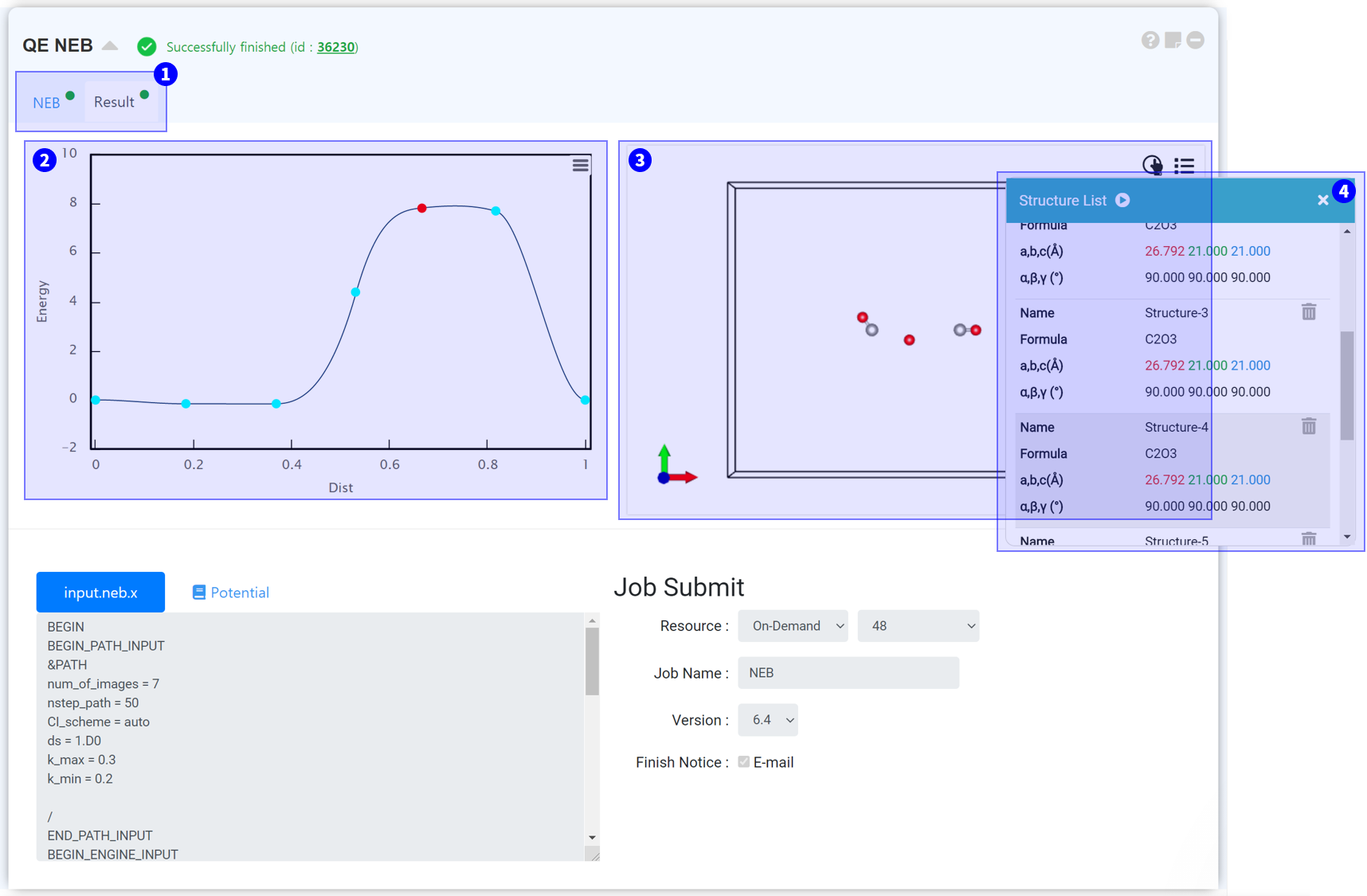
NEB 작업이 정상적으로 종료되면 Result 탭이 표시됩니다. Result 탭을 클릭하여 결과를 확인하십시오.
Reaction path 중 변화하는 에너지를 보여주는 그래프입니다.
Reaction path의 구조를 볼 수 있습니다.
③ Visualizer 우측 상단 Structure list icon을 클릭하면 structure list를 띄울 수 있습니다. 여기서 각 path의 구조를 확인할 수 있으며, 재생 버튼을 누르면 Reaction path를 애니메이션 형태로 볼 수 있습니다.
NEB 계산을 위해 Reaction Path (NEB) 모듈에 연결된 structure builder의 structrue list에 initial, final image (혹은 필요하다면 intermediate image까지)를 추가해 두어야 합니다. 더 나은 결과를 위해 모든 image는 미리 구조 최적화 (structual relaxation/optimization)가 완료된 구조를 사용하는 것이 좋습니다.
NEB Input script 설정에 대한 자세한 정보는 Quantum Espresso Input description (neb.x) 을 참고하십시오.
Reaction Path (NEB) 모듈은 General과 Manual 모드를 선택할 수 있습니다.
원자를 고정할 때 쓰는 도구입니다. 연결된 Structure builder의 structure list에 있는 구조의 순서대로 탭이 나뉘어 있습니다. 원하는 원자를 선택하면 해당 원자는 계산 중 고정된 것으로 간주됩니다.
neb.x의 모든 키워드 목록을 볼 수 있는 페이지입니다.
&PATH namelist에 속하는 키워드는 NEB 계산의 조건을 설정하는 키워드입니다. ③ Keyword information을 참고하여 적절하게 설정하십시오. Climbing 키워드를 'auto' 로 설정하면 Climbing NEB 계산을 수행할 수 있습니다.
NEB 경로를 결정할 때 사용되는 키워드입니다. 구조 Relaxation 시 사용한 input script를 복사하여 붙여넣을 수 있습니다.
설정한 input parameter는 input script 형태로 변환되어 서버에 전송됩니다. 잘못된 점은 없는지 확인하십시오.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
NEB 작업이 정상적으로 종료되면 Result 탭이 표시됩니다. Result 탭을 클릭하여 결과를 확인하십시오.
Reaction path 중 변화하는 에너지를 보여주는 그래프입니다.
Reaction path의 구조를 볼 수 있습니다.
③ Visualizer 우측 상단 Structure list icon을 클릭하면 structure list를 띄울 수 있습니다. 여기서 각 path의 구조를 확인할 수 있으며, 재생 버튼을 누르면 Reaction path를 애니메이션 형태로 볼 수 있습니다.
'Phonon' 은 Quantum Espresso에서 제공되는 ph.x solver를 이용하여 포논 계산을 수행하기 위한 모듈입니다.
포논 계산은 원자 간의 진동 (vibration) 을 기술하여야 하므로 매우 '안정된 구조' 로부터 시작하여야 하며, 계산 역시 매우 '정밀한' 계산을 수행하여야 합니다.
다음과 같은 절차로 포논 계산을 수행하는 것이 좋습니다.
1. Primitive cell에 대해 높은 정확도로 (vc-)relax 계산
2. Step 1의 Quantum Espresso module에 연결하여 Phonon 계산
3. 결과 후처리
보다 자세한 정보는 [MatSQ Tip] Phonon의 정의와 계산 수행, [MatSQ Tip] Phonon Dispersion 결과의 신뢰도를 판단하는 방법 및 계산 예제, Phonon 웨비나 비디오를 참고하십시오.
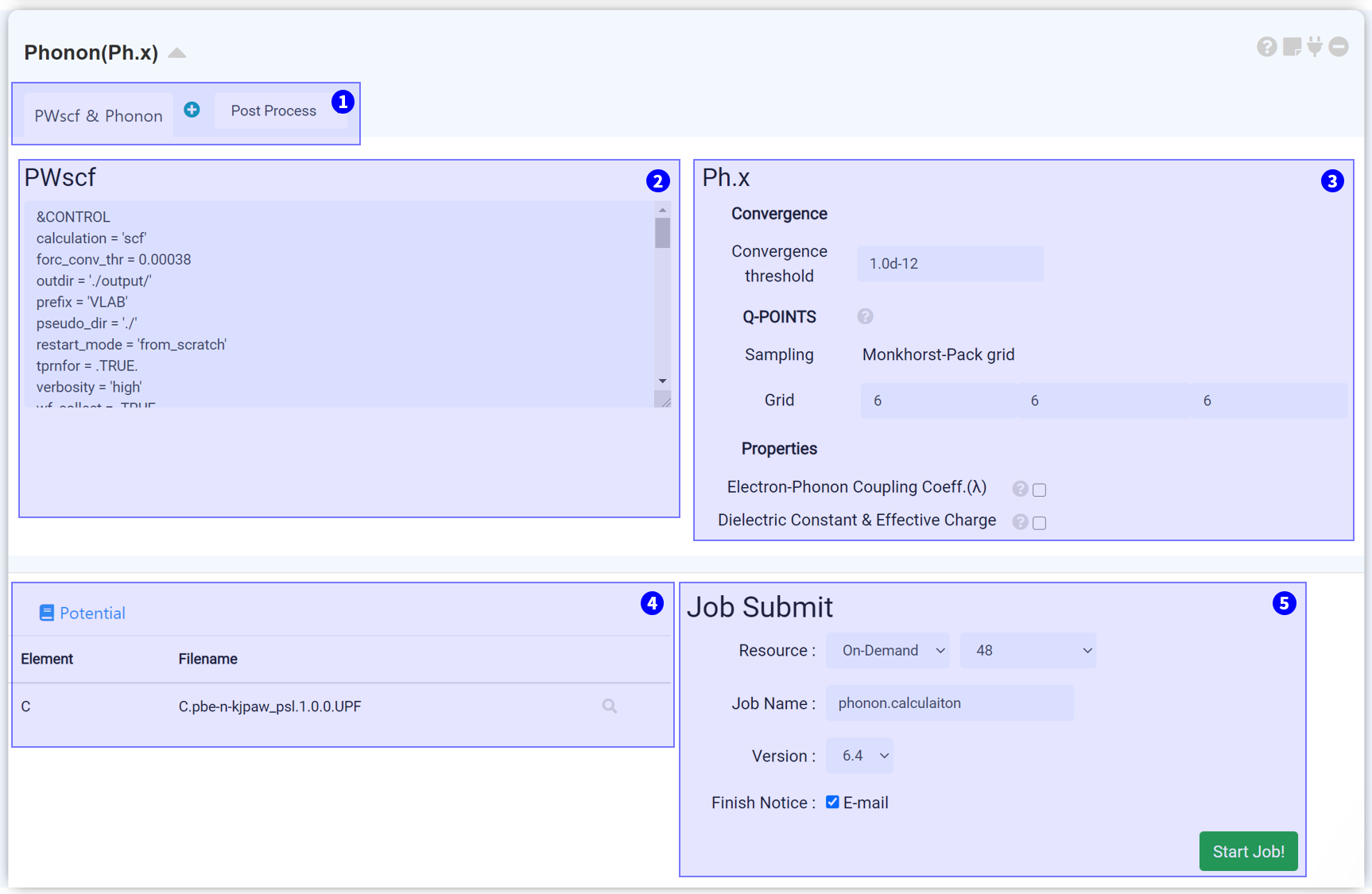
Post process 탭을 추가하여 Phonon 계산으로부터 Band structure 및 DOS 그래프를 얻을 수 있습니다. 자세한 설명은 다음 Post process 설정 문서를 참고하십시오.
SCF 계산을 위한 input script입니다. 기본적으로, Phonon 모듈과 연결한 Quantum Espresso 모듈의 input script가 복사되지만 Calculation type은 scf로 변경됩니다.
Phonon 계산에서의 수렴 한계를 결정하십시오. 기본값은 1.0d-12 Ry이며, 결과에 직접적으로 영향을 미치므로 값을 변경할 때 주의할 필요가 있습니다.
Phonon 계산에 사용되는 q-point를 설정합니다. k-point와 같은 방식으로 설정하여야 하나, k-point와 동일한 값으로 설정하면 계산량이 매우 많아질 수 있으므로 k-point보다 작은 grid로 설정하십시오.
Phonon 계산을 통해 얻을 특성을 결정합니다. Electron-Phonon Coupling Coefficient (λ) 는 금속 시스템 (occupation = "smearing"), Dielectric Constant & Effective Charge는 비금속 시스템 (occupation = "fixed")에 대해서만 계산할 수 있습니다.
계산에 사용될 pseudopotential을 설정합니다. Restart 계산일 경우, 연결된 Quantum Espresso 모듈에서 사용한 파일과 동일하여야 합니다.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
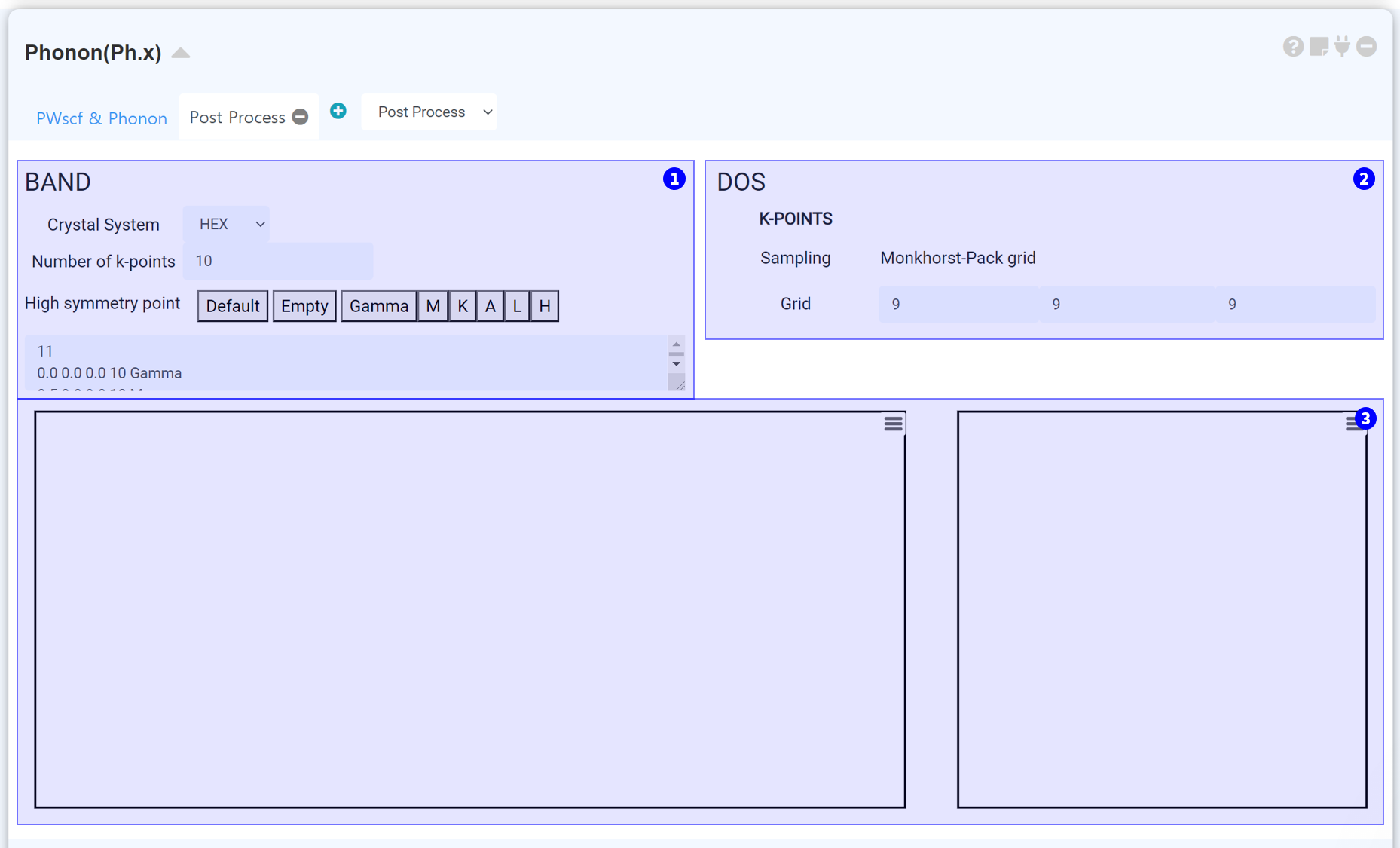
Phonon Band structure 계산에서 Band k-path를 설정합니다. 올바른 Crystal system을 선택하고, Number of k-point를 적절히 조절하십시오. Number of k-point 값을 크게 설정할수록 두 high symmetry point 사이에 샘플링하는 k-point의 수가 많아집니다.
Phonon DOS 계산에서의 k-point grid를 설정합니다.
계산이 성공적으로 종료되면, 이 부분에 Band structure와 DOS가 표시됩니다.
포논 계산은 원자 간의 진동 (vibration) 을 기술하여야 하므로 매우 '안정된 구조' 로부터 시작하여야 하며, 계산 역시 매우 '정밀한' 계산을 수행하여야 합니다.
다음과 같은 절차로 포논 계산을 수행하는 것이 좋습니다.
1. Primitive cell에 대해 높은 정확도로 (vc-)relax 계산
2. Step 1의 Quantum Espresso module에 연결하여 Phonon 계산
3. 결과 후처리
보다 자세한 정보는 [MatSQ Tip] Phonon의 정의와 계산 수행, [MatSQ Tip] Phonon Dispersion 결과의 신뢰도를 판단하는 방법 및 계산 예제, Phonon 웨비나 비디오를 참고하십시오.
Post process 탭을 추가하여 Phonon 계산으로부터 Band structure 및 DOS 그래프를 얻을 수 있습니다. 자세한 설명은 다음 Post process 설정 문서를 참고하십시오.
SCF 계산을 위한 input script입니다. 기본적으로, Phonon 모듈과 연결한 Quantum Espresso 모듈의 input script가 복사되지만 Calculation type은 scf로 변경됩니다.
Phonon 계산에서의 수렴 한계를 결정하십시오. 기본값은 1.0d-12 Ry이며, 결과에 직접적으로 영향을 미치므로 값을 변경할 때 주의할 필요가 있습니다.
Phonon 계산에 사용되는 q-point를 설정합니다. k-point와 같은 방식으로 설정하여야 하나, k-point와 동일한 값으로 설정하면 계산량이 매우 많아질 수 있으므로 k-point보다 작은 grid로 설정하십시오.
Phonon 계산을 통해 얻을 특성을 결정합니다. Electron-Phonon Coupling Coefficient (λ) 는 금속 시스템 (occupation = "smearing"), Dielectric Constant & Effective Charge는 비금속 시스템 (occupation = "fixed")에 대해서만 계산할 수 있습니다.
계산에 사용될 pseudopotential을 설정합니다. Restart 계산일 경우, 연결된 Quantum Espresso 모듈에서 사용한 파일과 동일하여야 합니다.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
Phonon Band structure 계산에서 Band k-path를 설정합니다. 올바른 Crystal system을 선택하고, Number of k-point를 적절히 조절하십시오. Number of k-point 값을 크게 설정할수록 두 high symmetry point 사이에 샘플링하는 k-point의 수가 많아집니다.
Phonon DOS 계산에서의 k-point grid를 설정합니다.
계산이 성공적으로 종료되면, 이 부분에 Band structure와 DOS가 표시됩니다.
High throughput 계산을 수행하기 위한 모듈입니다. Structure Builder에 연결한 시점에서 Structure list에 추가된 모든 구조 정보를 가져와 대량 계산 input script를 생성합니다.
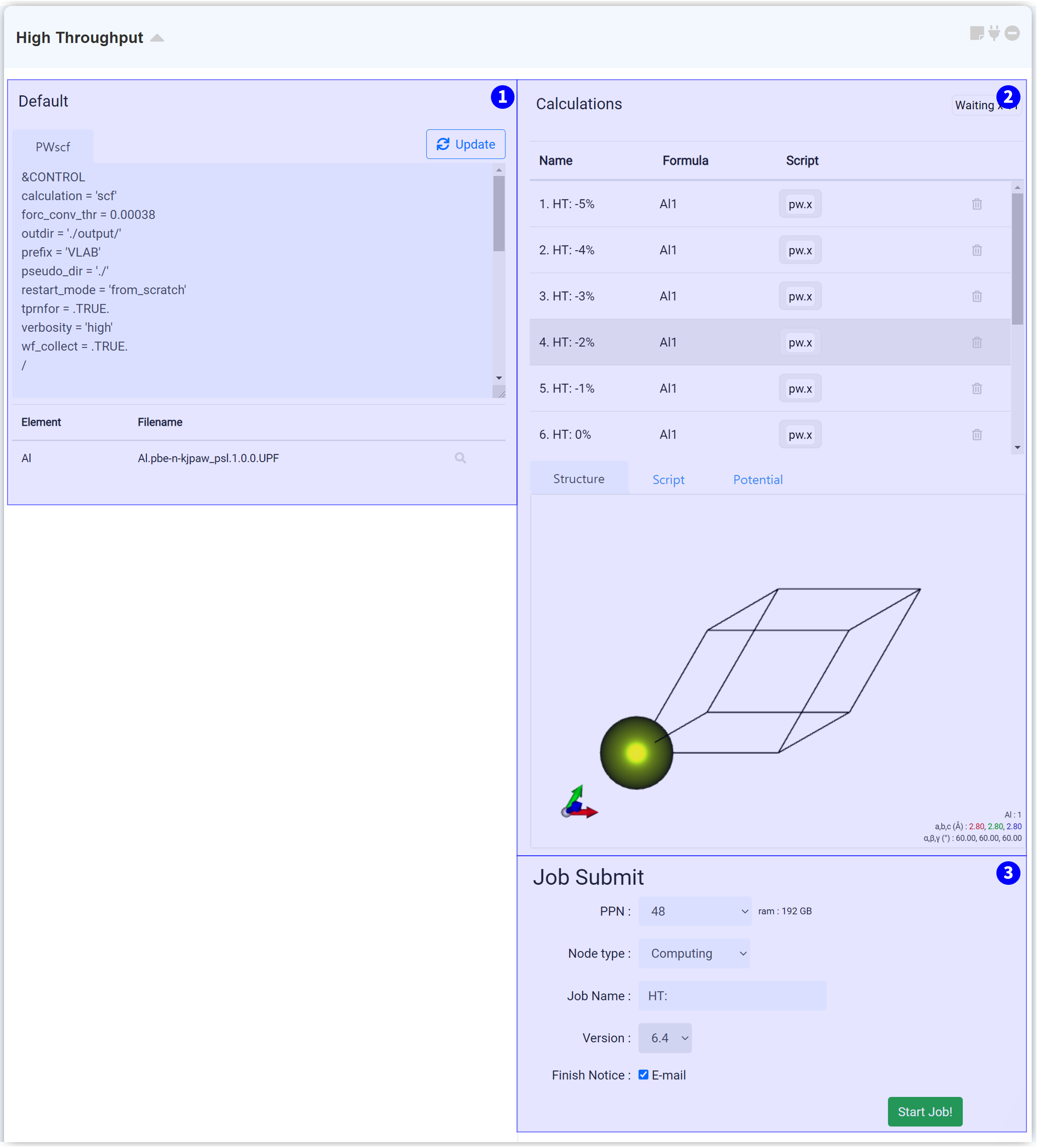
전체적인 input script와 pseudopotential을 수정하고 적용할 수 있습니다. 간편한 설정을 위해, Quantum Espresso (General) 모듈에서 input script를 설정한 다음 붙여넣으십시오.
원하는 Input script와 pseudopotential을 선택한 다음, 우측 상단의 ‘Update’ 버튼을 누르면 해당 모듈에 포함된 모든 high throughput 작업의 input들이 일괄 변경됩니다.
각각의 개별 작업을 변경할 수 있는 영역입니다. 해당 개별 작업의 계산 모델을 확인하고, input script와 pseudopotential을 개별적으로 변경할 수 있습니다.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
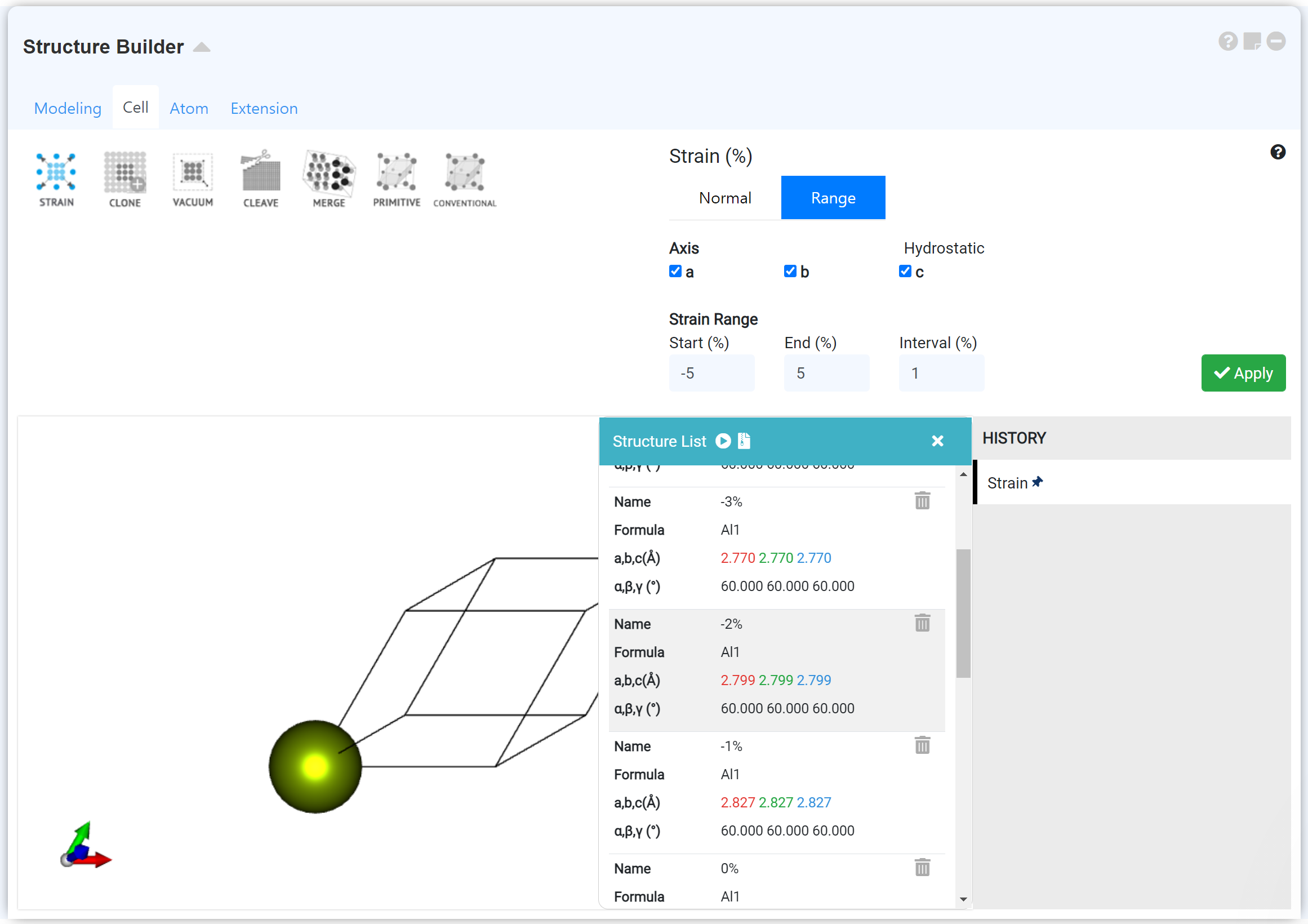
'Structure Builder' 모듈 'Cell' 탭의 'Strain (Range)' 메뉴를 이용해 부피가 변화하는 구조 세트를 모델링하여 equation of state를 계산할 수 있습니다. 자세한 내용은 Webinar - MatSQ 109: A New Paradigm of Materials Research-Machine Learning(CGCNN), [MatSQ Tip] High Throughput 모듈을 통한 Bulk Modulus 계산을 참고하십시오.
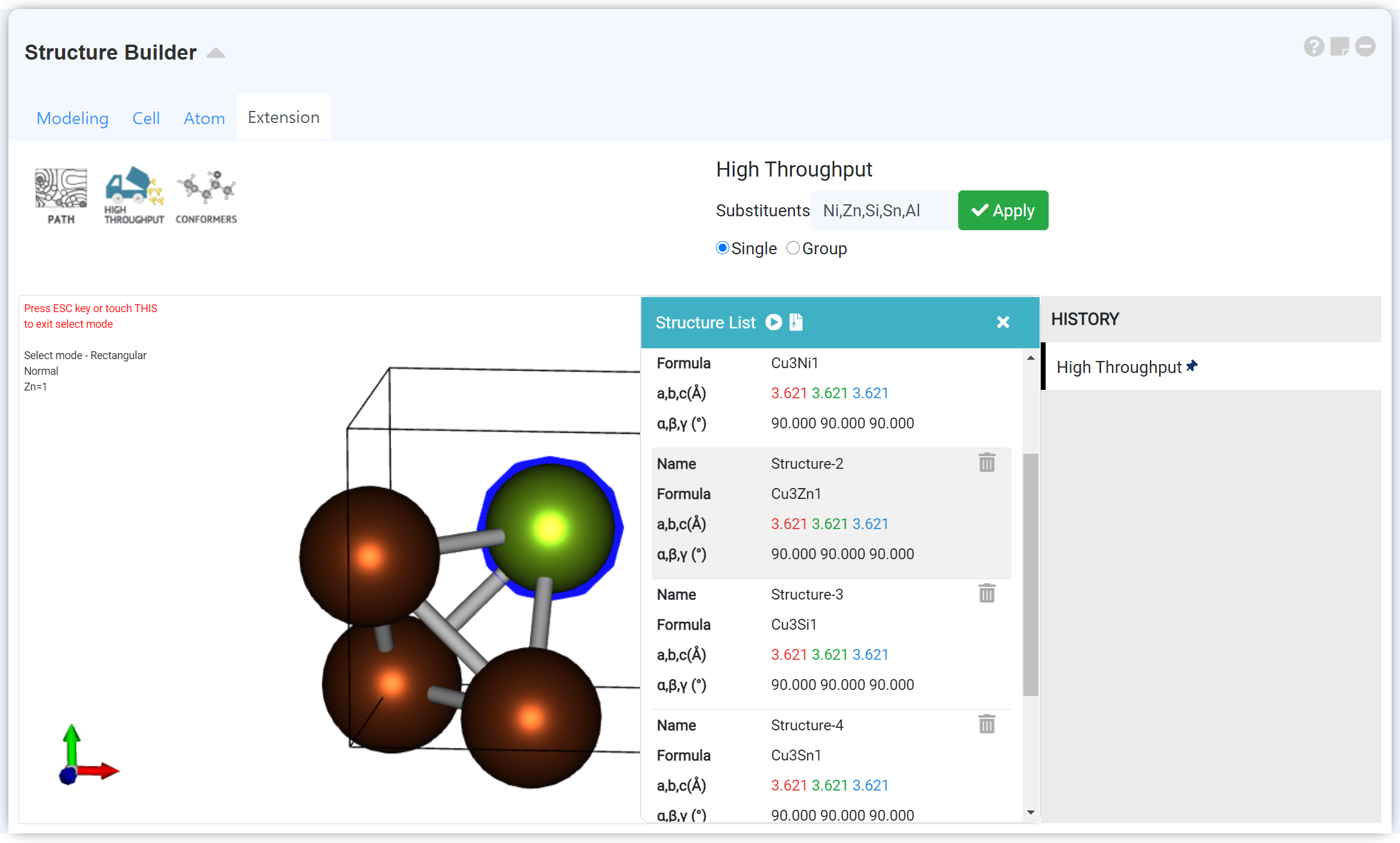
'Structure Builder' 모듈 'Extension' 탭의 'High Throughput' 메뉴를 사용하여 다양한 dopant로 치환된 구조 세트를 만들고, high throughput 모듈을 이용해 구조 최적화 계산 및 전자구조 특성 계산을 한 번에 수행할 수 있습니다. 자세한 내용은 'Structure Builder' 모듈 문서의 Manipulate the structure > Extension 탭을 참고하십시오.
전체적인 input script와 pseudopotential을 수정하고 적용할 수 있습니다. 간편한 설정을 위해, Quantum Espresso (General) 모듈에서 input script를 설정한 다음 붙여넣으십시오.
원하는 Input script와 pseudopotential을 선택한 다음, 우측 상단의 ‘Update’ 버튼을 누르면 해당 모듈에 포함된 모든 high throughput 작업의 input들이 일괄 변경됩니다.
각각의 개별 작업을 변경할 수 있는 영역입니다. 해당 개별 작업의 계산 모델을 확인하고, input script와 pseudopotential을 개별적으로 변경할 수 있습니다.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
'Structure Builder' 모듈 'Cell' 탭의 'Strain (Range)' 메뉴를 이용해 부피가 변화하는 구조 세트를 모델링하여 equation of state를 계산할 수 있습니다. 자세한 내용은 Webinar - MatSQ 109: A New Paradigm of Materials Research-Machine Learning(CGCNN), [MatSQ Tip] High Throughput 모듈을 통한 Bulk Modulus 계산을 참고하십시오.
'Structure Builder' 모듈 'Extension' 탭의 'High Throughput' 메뉴를 사용하여 다양한 dopant로 치환된 구조 세트를 만들고, high throughput 모듈을 이용해 구조 최적화 계산 및 전자구조 특성 계산을 한 번에 수행할 수 있습니다. 자세한 내용은 'Structure Builder' 모듈 문서의 Manipulate the structure > Extension 탭을 참고하십시오.
시뮬레이션 모듈의 SIESTA 모듈은 DFT 기반 계산 소프트웨어 패키지인 SIESTA를 이용하여 전자 구조 계산 (electronic structure calculation)을 수행하기 위한 모듈입니다.
Materials Square는 복잡한 코드 공부 없이 클릭 몇 번만으로 DFT 계산을 쉽게 수행할 수 있도록 직관적인 그래픽 인터페이스 (GUI)를 제공하고 있습니다.
대부분의 계산은 "General" 모드를 통해 Calculation type, Mesh Cutoff, K-point와 같은 주요 입력 변수를 바꾸는 것만으로 원하는 시뮬레이션 결과를 얻으실 수 있습니다. 숙련된 사용자를 위해 script를 직접 수정할 수 있는 "Manual" 모드도 제공하고 있습니다.
SIESTA에 대한 자세한 정보는 SIESTA 공식 홈페이지 에서 확인할 수 있습니다.
SIESTA를 통해 DFT 기반의 기본 계산을 실행할 수 있는 모듈입니다. 이 모듈에서 실행할 수 있는 계산은 1. 원자/전자 구조 최적화 계산, 2. Phonon 계산, 3. AIMD 계산 입니다. 각각의 계산은 "Calculation Type" 에서 선택할 수 있습니다.
1. 원자/전자 구조 최적화 계산
DFT 기반으로 물질의 성질을 얻기 위해 가장 먼저 수행되어야 하는 계산입니다. 시스템의 원자 위치나 전자 분포상태를 반복적으로 조정하여 시스템의 전체 에너지를 최소화되는 시스템의 원자 배열/전자 구성을 찾는 계산입니다. 시스템에 포함된 원자/전자의 수가 많고 수렴기준이 높을수록 많은 계산 리소스가 필요합니다.
2. Phonon 계산
포논 계산은 원자 간의 진동 (vibration) 을 기술하는 계산입니다. 이 계산은 매우 '안정된 구조' 로부터 시작하여야 하며, 매우 '정밀한' 계산을 수행하여야 합니다.
3. AIMD 계산
SIESTA 에서는 양자역학 기반의 분자 움직임을 모사하는 Ab initio Molecular Dynamics (AIMD) 계산을 제공합니다.
Classical MD와는 다르게 AIMD는 양자역학의 원리를 기반으로 하기 때문에 Classical MD보다 좀 더 정확한 분자의 물리적/화학적 특성 예측이 가능합니다. 다만, 전자 상호작용 또한 고려하여 계산하기 때문에 원자수의 제한이 있으며 classical MD보다 많은 계산 리소스가 필요합니다.
SIESTA에서 제공하는 MD 알고리즘은 아래와 같습니다.
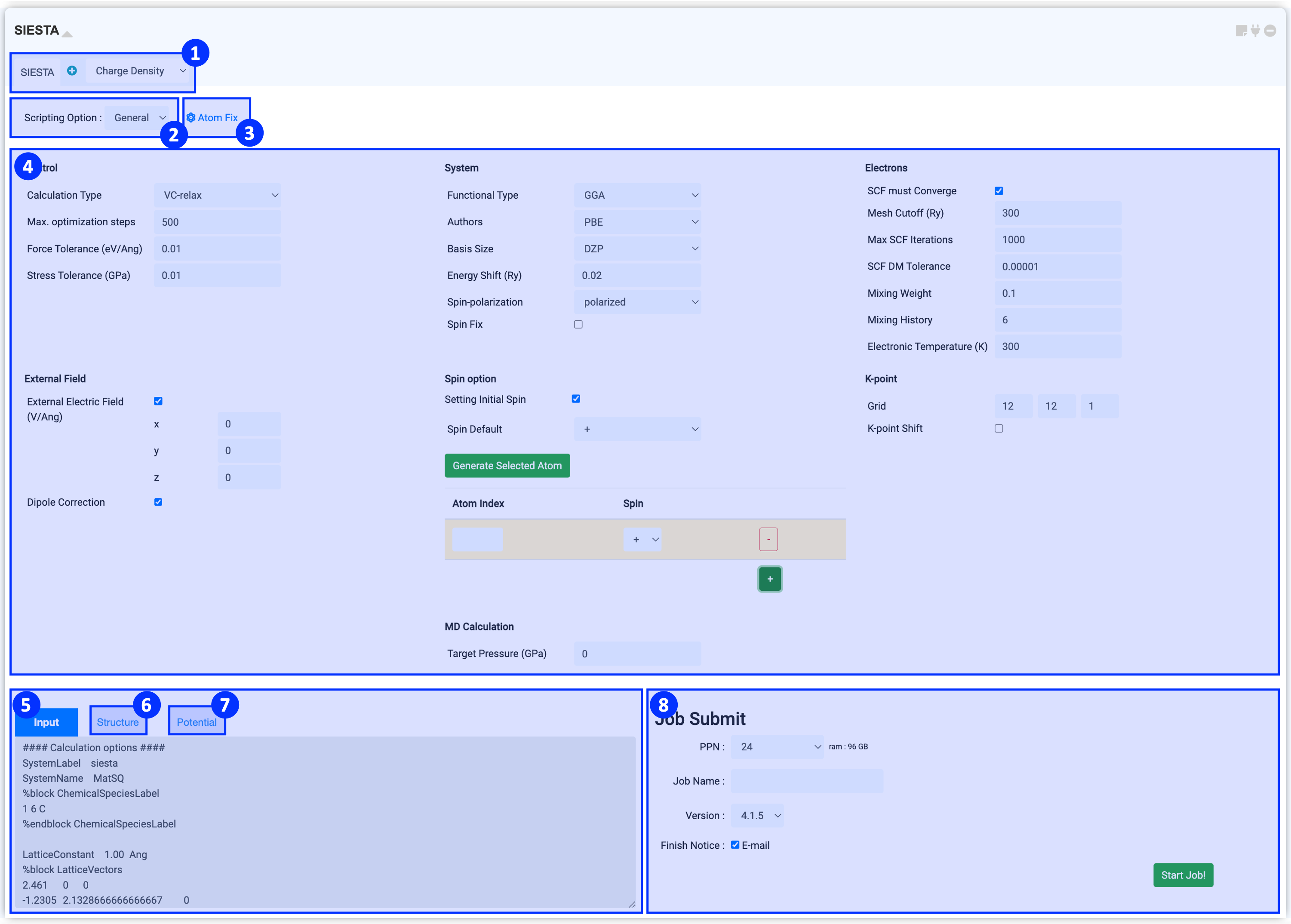
모듈 상단부에서는 계산을 위한 solver를 선택하실 수 있습니다. Solver에는 기본 계산을 실행할 수 있는 “"SIESTA"와 함께 후처리 계산을 위한 다양한 solver들을 확인할 수 있습니다.
SIESTA solver로 cell & atomic structure relaxation (vc-relax) , atomic structure relaxation (relax), electronic structure relaxation (scf), phonon calculation, ab initio molecular dynamics (AIMD) calculation이 있습니다. 후처리 solver 에 대한 부분은 Post-processing 계산 을 참고하십시오.
Scripting option을 누르면 General 과 Manual 옵션을 볼 수 있습니다. General 모드는 일반적으로 많이 사용하는 input keyword 에 대해 적절한 값으로 설정되어 있으며, 사용자가 더 많은 keyword 설정을 필요로 한다면 Manual 모드를 사용할 수 있습니다.
SIESTA 모듈은 연결된 structure builder에서 구조 정보를 가져옵니다. Atom Fix를 클릭하여 열린 창에서 원자들의 좌표값을 클릭하시면 선택된 해당 원자는 DFT 계산 중 위치가 고정됩니다. Structure Builder에서 고정하시고자 하는 원자를 선택하신 뒤 "selected"를 클릭하시면 손쉽게 원자 고정을 실행하실 수 있습니다.
Materials Square의 SIESTA 모듈에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있도록 General 모드를 구성하였습니다. General 모드에서는 기본적으로 자주 쓰이는 input keyword에 대해 일반적으로 적절한 값을 설정해두었습니다. 대부분의 계산은 주요한 keyword의 값을 수정하는 것만으로 합리적인 결과를 얻을 수 있지만, 논문 작성 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input keyword에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다. 각 계산 방식의 input parameter에 대한 자세한 정보는 Appendix 에서 확인할 수 있습니다.
SIESTA 모듈을 Structure Builder에 연결한 후 Input parameter를 설정하면 input script와 구조 정보가 자동으로 작성됩니다. ⑤ Input 에는 계산에 필요한 keyword 설정 정보를 갖고 있으며 구조와 관련된 정보는 ⑥ Structure에 작성됩니다. 이 정보는 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ④ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다.
DFT 계산을 수행할 때 해당 원자의 거동을 결정하는 것이 바로 pseudopotential입니다. 그렇기 때문에 계산 전에 먼저 사용하시려는 Psuedopotential 파일을 셋팅해야 합니다. Psuedopotential 파일 셋팅 방법은 "Pseudopotential setting" 을 참고하십시오.
⑦ Pseudopotential 탭에서는 셋팅된 파일을 보여줍니다. 돋보기 버튼을 누르면 사용할 수 있는 pseudopotential 리스트를 볼 수 있습니다. MatSQ database에는 pseudopotential file은 NNIN Virtual Vault for Pseudopotentials 에서 다운로드 받은 파일을 기본으로 사용하고 있습니다. Pseudopotential에 대한 자세한 정보는 Pseudopotential Files 를 참고하십시오.
①~⑦의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서 를 참고하여 계산 작업을 시작하십시오.
계산이 종료된 SIESTA 모듈에 새로운 SIESTA 모듈을 연결하면, 계산이 종료된 지점에서 계산을 이어 시작하는 restart 계산을 진행할 수 있습니다. 단, 오류가 발생한 계산으로부터는 재시작할 수 없습니다. 오류에 대한 설명은 "Trouble shooting" 을 참고하십시오.
Restart 계산은 전단계에서 실행한 계산에서 input parameter 정보와 마지막 step의 구조와를 불러와 계산을 이어 진행하는 것입니다. SIESTA 모듈을 기존 SIESTA 모듈에 연결하면 기존 모듈에서 setting한 input parameter와 최종 구조가 새로운 모듈로 복사 됩니다.
Restart 계산은 다음 경우에 적용하면 좋습니다.
1. "Structure is not relaxed" 메시지가 표시됐을 때 : 원자 구조 최적화과정에서 설정한 step수 (Max Iteration Step)만큼 계산이 진행되었지만 convergence force tolerance 값 안에 도달하지 못한 경우입니다. 이런 경우 최종 구조 및 input parameter 를 불러와서 restart 계산을 실행합니다. 동일한 input parameter를 사용하지만, 다시 최적화가 완료되지 않을 경우를 고려하여 Max Iteration Step 수를 늘려 재계산하는 것이 좋습니다.
2. Post-processing 계산을 진행할 때 : 이 계산은 최적화가 완료된 원자구조로부터 전자구조 분석을 위한 post-processing 계산을 위해 restart를 수행하는 것입니다. DOS, Band Structures, Optical Property 계산은Calculation Type을 SCF로 변경하고 분석을 원하는 post-processing solver를 추가하면 됩니다.
후처리 계산은 "SIESTA" solver를 통한 계산이 완료 된 후 수행되며, 계산을 원하는 후처리 solver를 추가하여 얻으실 수 있습니다. 후처리 계산 solver를 추가하는 방법은 아래와 같습니다.
Materials Square는 복잡한 코드 공부 없이 클릭 몇 번만으로 DFT 계산을 쉽게 수행할 수 있도록 직관적인 그래픽 인터페이스 (GUI)를 제공하고 있습니다.
대부분의 계산은 "General" 모드를 통해 Calculation type, Mesh Cutoff, K-point와 같은 주요 입력 변수를 바꾸는 것만으로 원하는 시뮬레이션 결과를 얻으실 수 있습니다. 숙련된 사용자를 위해 script를 직접 수정할 수 있는 "Manual" 모드도 제공하고 있습니다.
SIESTA에 대한 자세한 정보는 SIESTA 공식 홈페이지 에서 확인할 수 있습니다.
SIESTA를 통해 DFT 기반의 기본 계산을 실행할 수 있는 모듈입니다. 이 모듈에서 실행할 수 있는 계산은 1. 원자/전자 구조 최적화 계산, 2. Phonon 계산, 3. AIMD 계산 입니다. 각각의 계산은 "Calculation Type" 에서 선택할 수 있습니다.
1. 원자/전자 구조 최적화 계산
DFT 기반으로 물질의 성질을 얻기 위해 가장 먼저 수행되어야 하는 계산입니다. 시스템의 원자 위치나 전자 분포상태를 반복적으로 조정하여 시스템의 전체 에너지를 최소화되는 시스템의 원자 배열/전자 구성을 찾는 계산입니다. 시스템에 포함된 원자/전자의 수가 많고 수렴기준이 높을수록 많은 계산 리소스가 필요합니다.
2. Phonon 계산
포논 계산은 원자 간의 진동 (vibration) 을 기술하는 계산입니다. 이 계산은 매우 '안정된 구조' 로부터 시작하여야 하며, 매우 '정밀한' 계산을 수행하여야 합니다.
3. AIMD 계산
SIESTA 에서는 양자역학 기반의 분자 움직임을 모사하는 Ab initio Molecular Dynamics (AIMD) 계산을 제공합니다.
Classical MD와는 다르게 AIMD는 양자역학의 원리를 기반으로 하기 때문에 Classical MD보다 좀 더 정확한 분자의 물리적/화학적 특성 예측이 가능합니다. 다만, 전자 상호작용 또한 고려하여 계산하기 때문에 원자수의 제한이 있으며 classical MD보다 많은 계산 리소스가 필요합니다.
SIESTA에서 제공하는 MD 알고리즘은 아래와 같습니다.
-
(1) Verlet (NVE): Standard Verlet algorithm MD
(2) Nose (NVT): temperature controlled by means of a Nosé thermostat
(3) ParrinelloRahman (NPE): pressure controlled by the Parrinello-Rahman method
(4) Nose-ParrinelloRahman (NPT): annealing to a desired temperature and/or pressure
(5) Anneal: annealing to a desired temperature and/or pressure
모듈 상단부에서는 계산을 위한 solver를 선택하실 수 있습니다. Solver에는 기본 계산을 실행할 수 있는 “"SIESTA"와 함께 후처리 계산을 위한 다양한 solver들을 확인할 수 있습니다.
SIESTA solver로 cell & atomic structure relaxation (vc-relax) , atomic structure relaxation (relax), electronic structure relaxation (scf), phonon calculation, ab initio molecular dynamics (AIMD) calculation이 있습니다. 후처리 solver 에 대한 부분은 Post-processing 계산 을 참고하십시오.
Scripting option을 누르면 General 과 Manual 옵션을 볼 수 있습니다. General 모드는 일반적으로 많이 사용하는 input keyword 에 대해 적절한 값으로 설정되어 있으며, 사용자가 더 많은 keyword 설정을 필요로 한다면 Manual 모드를 사용할 수 있습니다.
SIESTA 모듈은 연결된 structure builder에서 구조 정보를 가져옵니다. Atom Fix를 클릭하여 열린 창에서 원자들의 좌표값을 클릭하시면 선택된 해당 원자는 DFT 계산 중 위치가 고정됩니다. Structure Builder에서 고정하시고자 하는 원자를 선택하신 뒤 "selected"를 클릭하시면 손쉽게 원자 고정을 실행하실 수 있습니다.
Materials Square의 SIESTA 모듈에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있도록 General 모드를 구성하였습니다. General 모드에서는 기본적으로 자주 쓰이는 input keyword에 대해 일반적으로 적절한 값을 설정해두었습니다. 대부분의 계산은 주요한 keyword의 값을 수정하는 것만으로 합리적인 결과를 얻을 수 있지만, 논문 작성 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input keyword에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다. 각 계산 방식의 input parameter에 대한 자세한 정보는 Appendix 에서 확인할 수 있습니다.
SIESTA 모듈을 Structure Builder에 연결한 후 Input parameter를 설정하면 input script와 구조 정보가 자동으로 작성됩니다. ⑤ Input 에는 계산에 필요한 keyword 설정 정보를 갖고 있으며 구조와 관련된 정보는 ⑥ Structure에 작성됩니다. 이 정보는 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ④ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다.
DFT 계산을 수행할 때 해당 원자의 거동을 결정하는 것이 바로 pseudopotential입니다. 그렇기 때문에 계산 전에 먼저 사용하시려는 Psuedopotential 파일을 셋팅해야 합니다. Psuedopotential 파일 셋팅 방법은 "Pseudopotential setting" 을 참고하십시오.
⑦ Pseudopotential 탭에서는 셋팅된 파일을 보여줍니다. 돋보기 버튼을 누르면 사용할 수 있는 pseudopotential 리스트를 볼 수 있습니다. MatSQ database에는 pseudopotential file은 NNIN Virtual Vault for Pseudopotentials 에서 다운로드 받은 파일을 기본으로 사용하고 있습니다. Pseudopotential에 대한 자세한 정보는 Pseudopotential Files 를 참고하십시오.
①~⑦의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서 를 참고하여 계산 작업을 시작하십시오.
계산이 종료된 SIESTA 모듈에 새로운 SIESTA 모듈을 연결하면, 계산이 종료된 지점에서 계산을 이어 시작하는 restart 계산을 진행할 수 있습니다. 단, 오류가 발생한 계산으로부터는 재시작할 수 없습니다. 오류에 대한 설명은 "Trouble shooting" 을 참고하십시오.
Restart 계산은 전단계에서 실행한 계산에서 input parameter 정보와 마지막 step의 구조와를 불러와 계산을 이어 진행하는 것입니다. SIESTA 모듈을 기존 SIESTA 모듈에 연결하면 기존 모듈에서 setting한 input parameter와 최종 구조가 새로운 모듈로 복사 됩니다.
Restart 계산은 다음 경우에 적용하면 좋습니다.
- "Structure is not relaxed" 메시지가 표시됐을 때
- DOS, Bands, optical, phonon 등 post-processing 계산을 진행할 때
1. "Structure is not relaxed" 메시지가 표시됐을 때 : 원자 구조 최적화과정에서 설정한 step수 (Max Iteration Step)만큼 계산이 진행되었지만 convergence force tolerance 값 안에 도달하지 못한 경우입니다. 이런 경우 최종 구조 및 input parameter 를 불러와서 restart 계산을 실행합니다. 동일한 input parameter를 사용하지만, 다시 최적화가 완료되지 않을 경우를 고려하여 Max Iteration Step 수를 늘려 재계산하는 것이 좋습니다.
2. Post-processing 계산을 진행할 때 : 이 계산은 최적화가 완료된 원자구조로부터 전자구조 분석을 위한 post-processing 계산을 위해 restart를 수행하는 것입니다. DOS, Band Structures, Optical Property 계산은Calculation Type을 SCF로 변경하고 분석을 원하는 post-processing solver를 추가하면 됩니다.
후처리 계산은 "SIESTA" solver를 통한 계산이 완료 된 후 수행되며, 계산을 원하는 후처리 solver를 추가하여 얻으실 수 있습니다. 후처리 계산 solver를 추가하는 방법은 아래와 같습니다.
- SIESTA 모듈을 추가하고, ① Solver에서 원하는 데이터를 얻기 위한 solver (DOS, charge density, 등)를 선택하십시오. 다음으로 + 버튼을 눌러 SIESTA 옆에 새 solver 탭을 추가합니다. Post-processing solver는 계산할 후처리 수만큼 추가할 수 있습니다.
- Phonon 계산의 경우, 자동으로 Phonon post-processing tab이 추가됩니다.
- Start Job! 버튼을 누르면 SIESTA 계산과 함께 추가된 모든 post-processing 작업들이 실행됩니다.
SIESTA 모듈의 potential file은 기본적으로 Norm-Conserving PP을 사용하며 MatSQ에서는 잘 알려진 CA(=PZ)와 PBE functional 에 대한 potential 파일을 제공하고 있습니다.
다만, 해당 potential file은 사용자의 특정한 시스템에 대한 적합성에 대한 책임을 지지 않습니다.
의사 전위와 기저 세트는 실제 문제에서 사용하기 전에 매우 잘 알려진 상황에서 테스트하고 사용하시길 추천 드립니다. 특히, 다른 방식의 potential type을 사용할 경우, 개인적으로 만드신 파일을 업로드하신 후 반드시 테스트 후 사용하시기 바랍니다.
의사 전위와 기저 세트는 실제 문제에서 사용하기 전에 매우 잘 알려진 상황에서 테스트하고 사용하시길 추천 드립니다. 특히, 다른 방식의 potential type을 사용할 경우, 개인적으로 만드신 파일을 업로드하신 후 반드시 테스트 후 사용하시기 바랍니다.
- Virtual Vault : SIESTA homepage에서 non-academic user들에게 추천한 Virtual Vault Database 에서 다운로드 받은 potential files 입니다.
- MatSQ : MatSQ Team이 ATOM 을 사용하여 만든 relativistic potential files입니다. Spin Orbit Coupling (SOC) 계산을 위해서는 반드시 이 potential file을 사용하거나 직접 upload한 relativistic file을 사용하여야 합니다.
- 사용자 업로드: 사용자가 만든 potential을 upload하여 사용할 수 있습니다. 이 때 upload 한 파일이 semicore가 설정된 potential file일 경우, input file에 %block PAO.Basis 를 수동으로 설정하여야 합니다.
GAMESS 모듈은 계산화학 오픈 소스 소프트웨어인 GAMESS(US)를 이용하여 전자 구조 계산 (electronic structure calculation)을 수행하기 위한 모듈입니다. MatSQ는 복잡한 코드 공부 없이 클릭 몇 번만으로 DFT 계산을 쉽게 수행할 수 있도록 직관적인 그래픽 인터페이스 (GUI)를 제공하고 있습니다. 특히, Scripting option: Template을 이용하면 버튼 몇 개를 클릭하는 것만으로도 input script를 작성할 수 있습니다. 이외에 숙련된 사용자를 위해 script를 직접 수정할 수 있는 manual 모드도 제공하고 있습니다.
GAMESS에 대한 자세한 정보는 www.msg.chem.iastate.edu/gamess/ 에서 확인할 수 있습니다. GAMESS Input parameter에 대한 자세한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
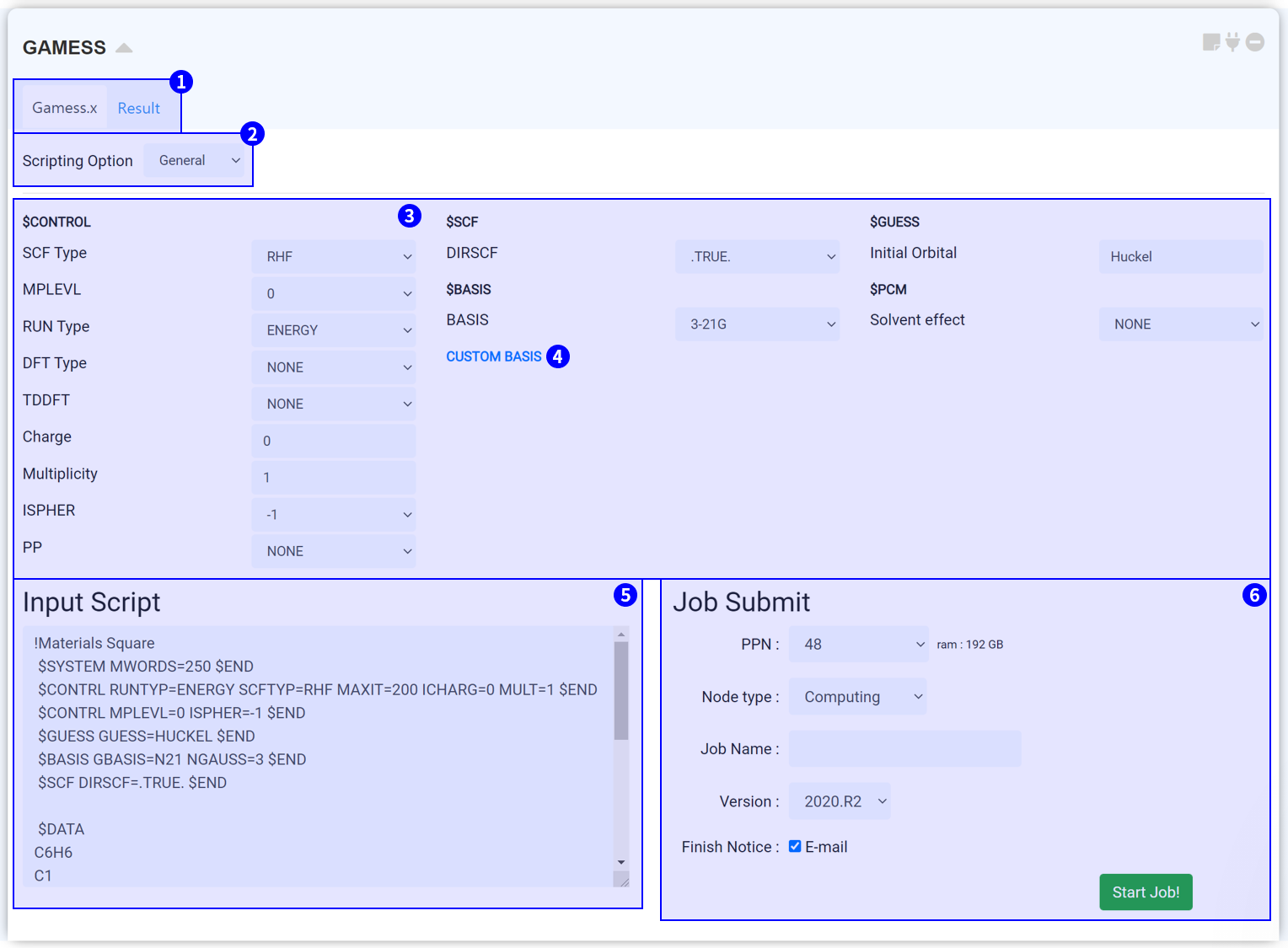
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
Scripting option을 누르면 Template, General, Manual 옵션을 볼 수 있습니다. Template은 몇 번의 클릭만을 통해 GAMESS input script를 설정할 수 있습니다. Template에서는 세 가지 질문에 답하여 input script를 설정합니다.
MatSQ에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있습니다. GAMESS 모듈에 기본적으로 설정된 input parameter는 일반적으로 적절한 값을 설정해 둔 값으로, 대부분의 계산은 원하는 특성을 위한 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 그러나 논문 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input parameter에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다. Input parameter에 대한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
GAMESS에서는 모든 원소에 대한 basis를 제공하지 않습니다. 특히 무거운 원소는 적절한 기본 제공 basis가 없는 경우가 많습니다. 그러한 경우, custom basis 파일을 찾아 적용해야 합니다. Custom basis는 Basis Set Exchange에서 확인할 수 있습니다.
GAMESS 모듈을 structure builder에 연결하고 Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ⑤ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다. Input script 창에서 텍스트를 수정하면 자동으로 Manual 모드로 변경됩니다.
①~⑤의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
계산이 종료된 GAMESS 모듈에 새로운 GAMESS 모듈을 연결하면, 계산이 종료된 지점에서 계산을 이어 시작하는 restart 계산을 진행할 수 있습니다. 특히 사용자가 임의로 중단한 계산을 되살리고 싶을 때 restart를 통해 계산을 이어 시작할 수 있습니다. 단, 오류가 발생한 계산으로부터는 재시작할 수 없습니다.
GAMESS에 대한 자세한 정보는 www.msg.chem.iastate.edu/gamess/ 에서 확인할 수 있습니다. GAMESS Input parameter에 대한 자세한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
Scripting option을 누르면 Template, General, Manual 옵션을 볼 수 있습니다. Template은 몇 번의 클릭만을 통해 GAMESS input script를 설정할 수 있습니다. Template에서는 세 가지 질문에 답하여 input script를 설정합니다.
- Target property: 어떠한 데이터를 얻고 싶은지 선택하십시오. 선택에 맞추어 적절한 input script가 설정됩니다.
- Energy/Optimization/Vibration: 계산 시 구조 최적화 여부 및 진동 특성 계산 여부를 결정합니다.
- Option: 계산에 어떠한 추가 옵션을 고려할 것인지 결정합니다.
MatSQ에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있습니다. GAMESS 모듈에 기본적으로 설정된 input parameter는 일반적으로 적절한 값을 설정해 둔 값으로, 대부분의 계산은 원하는 특성을 위한 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 그러나 논문 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input parameter에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다. Input parameter에 대한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
GAMESS에서는 모든 원소에 대한 basis를 제공하지 않습니다. 특히 무거운 원소는 적절한 기본 제공 basis가 없는 경우가 많습니다. 그러한 경우, custom basis 파일을 찾아 적용해야 합니다. Custom basis는 Basis Set Exchange에서 확인할 수 있습니다.
GAMESS 모듈을 structure builder에 연결하고 Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ⑤ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다. Input script 창에서 텍스트를 수정하면 자동으로 Manual 모드로 변경됩니다.
①~⑤의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
계산이 종료된 GAMESS 모듈에 새로운 GAMESS 모듈을 연결하면, 계산이 종료된 지점에서 계산을 이어 시작하는 restart 계산을 진행할 수 있습니다. 특히 사용자가 임의로 중단한 계산을 되살리고 싶을 때 restart를 통해 계산을 이어 시작할 수 있습니다. 단, 오류가 발생한 계산으로부터는 재시작할 수 없습니다.
GAMESS-IRC 모듈은 계산화학 오픈 소스 소프트웨어인 GAMESS(US)를 이용하여 IRC (Intrinsic reaction coordinate) 계산을 수행하기 위한 모듈입니다.
하나의 허수 진동수 (안장점, saddle point)가 존재하는 GAMESS 계산에 연결해야 합니다. 즉 GAMESS에서 ‘RUNTYP=SADPOINT’ 계산을 먼저 실행해야 합니다.
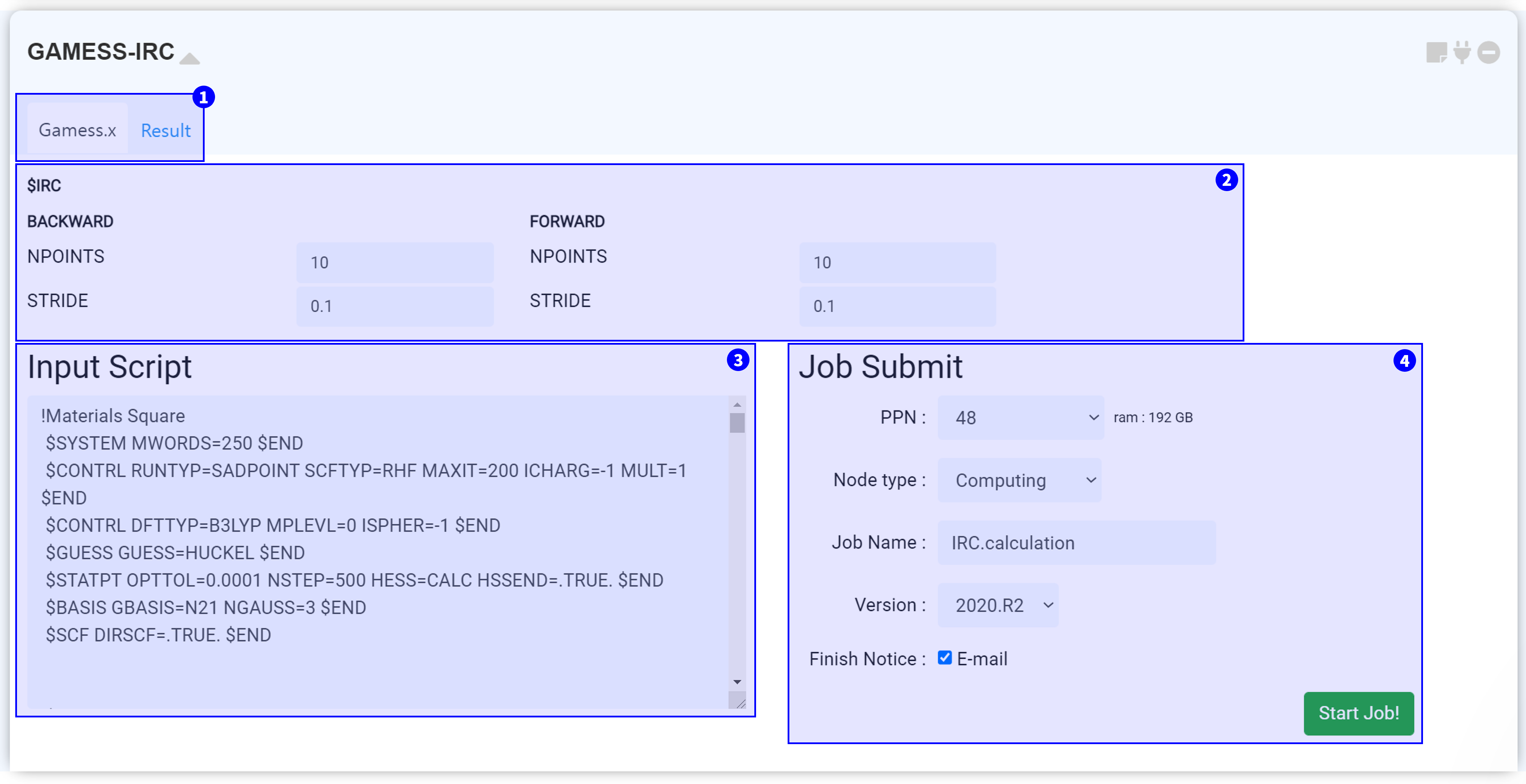
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS-IRC 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
MatSQ에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있습니다. GAMESS 모듈에 기본적으로 설정된 input parameter는 일반적으로 적절한 값을 설정해 둔 값으로, 대부분의 계산은 원하는 특성을 위한 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 그러나 논문 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input parameter에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다. GAMESS Input parameter에 대한 자세한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
GAMESS-IRC 모듈을 안장점 계산을 수행한 GAMESS 모듈에 연결하고 ② Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다.
①~③의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
하나의 허수 진동수 (안장점, saddle point)가 존재하는 GAMESS 계산에 연결해야 합니다. 즉 GAMESS에서 ‘RUNTYP=SADPOINT’ 계산을 먼저 실행해야 합니다.
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS-IRC 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
MatSQ에서는 단 몇 번의 클릭만으로 DFT 계산을 쉽게 수행할 수 있습니다. GAMESS 모듈에 기본적으로 설정된 input parameter는 일반적으로 적절한 값을 설정해 둔 값으로, 대부분의 계산은 원하는 특성을 위한 주요한 키워드를 바꾸는 것만으로 괜찮은 결과를 얻을 수 있습니다. 그러나 논문 등 연구에 이용하기 위해 신뢰도 높은 DFT 시뮬레이션을 수행하기 위해서는 각 Input parameter에 대한 이해를 바탕으로 값을 신중히 고려하여 선택해야 합니다. GAMESS Input parameter에 대한 자세한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
GAMESS-IRC 모듈을 안장점 계산을 수행한 GAMESS 모듈에 연결하고 ② Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다.
①~③의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
GAMESS-NEB 모듈은 계산화학 오픈 소스 소프트웨어인 GAMESS(US)를 이용하여 Nudged Elastic Band (NEB) 계산을 수행하기 위한 모듈입니다.
NEB 계산을 위해서는 2개 이상의 구조를 모델링한 Structure Builder에 연결해야 합니다. Structure Builder의 Structure list에 Initial, Intermediate (optional), Final 구조를 추가하십시오. 모든 이미지는 먼저 최적화를 진행하는 것이 좋습니다.
이때, 모든 분자의 원자 순서가 동일해야 합니다. 원자 순서는 ‘Edit’ 메뉴에서 확인할 수 있습니다.
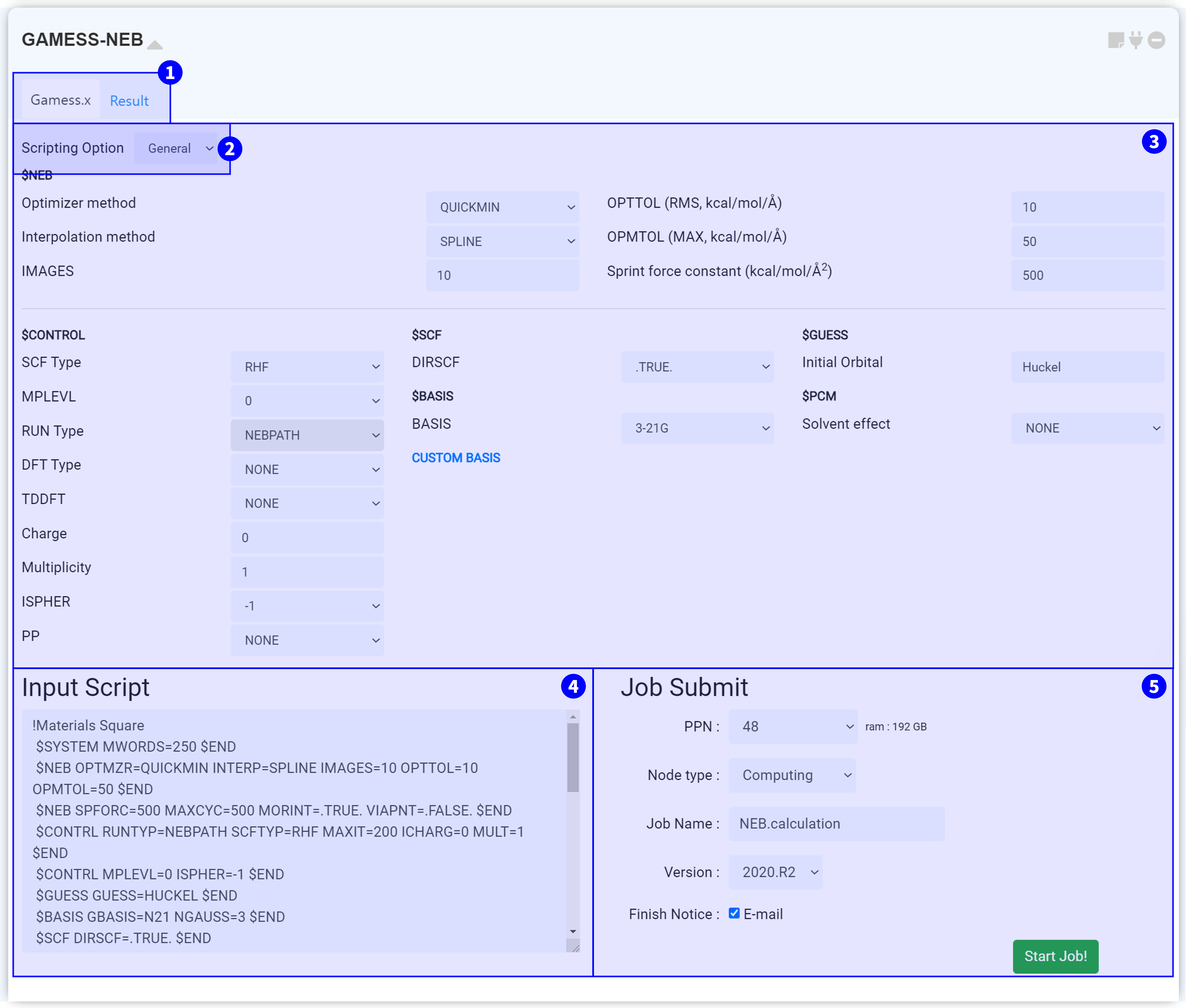
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS-NEB 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
Scripting option을 누르면 General, Manual 옵션을 볼 수 있습니다.
GAMESS-NEB 모듈의 Input 설정창은 크게 두 부분으로 나눌 수 있습니다. $NEB 그룹에서는 NEB 계산 옵션을 수정할 수 있으며, $CONTROL을 포함한 다른 그룹에서는 수렴 옵션을 조절할 수 있습니다. GAMESS Input parameter에 대한 자세한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
GAMESS 모듈을 structure builder에 연결하고 Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ③ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다. Input script 창에서 텍스트를 수정하면 자동으로 Manual 모드로 변경됩니다.
①~④의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
NEB 계산을 위해서는 2개 이상의 구조를 모델링한 Structure Builder에 연결해야 합니다. Structure Builder의 Structure list에 Initial, Intermediate (optional), Final 구조를 추가하십시오. 모든 이미지는 먼저 최적화를 진행하는 것이 좋습니다.
이때, 모든 분자의 원자 순서가 동일해야 합니다. 원자 순서는 ‘Edit’ 메뉴에서 확인할 수 있습니다.
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS-NEB 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
Scripting option을 누르면 General, Manual 옵션을 볼 수 있습니다.
GAMESS-NEB 모듈의 Input 설정창은 크게 두 부분으로 나눌 수 있습니다. $NEB 그룹에서는 NEB 계산 옵션을 수정할 수 있으며, $CONTROL을 포함한 다른 그룹에서는 수렴 옵션을 조절할 수 있습니다. GAMESS Input parameter에 대한 자세한 설명은 GAMESS Manual 또는 GAMESS Input 알아보기를 참고하십시오.
GAMESS 모듈을 structure builder에 연결하고 Input parameter를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다. ② Scripting Option에서 옵션을 Manual로 변경하면 ③ Input parameters 부분이 생략되고 Input script 창만 남게 되며, script를 직접 수정할 수 있습니다. Input script 창에서 텍스트를 수정하면 자동으로 Manual 모드로 변경됩니다.
①~④의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
GAMESS-BDE 모듈은 계산화학 오픈 소스 소프트웨어인 GAMESS(US)를 이용하여 Bond Dissociation Energy (BDE) 계산을 수행하기 위한 모듈입니다. GAMESS-BDE 모듈은 구조 최적화 계산을 완료한 GAMESS 모듈에 연결해야 합니다. 에너지뿐만 아니라 열역학 정보까지 보려면 Hessian 계산을 수행한 GAMESS 모듈에 연결해야 합니다.
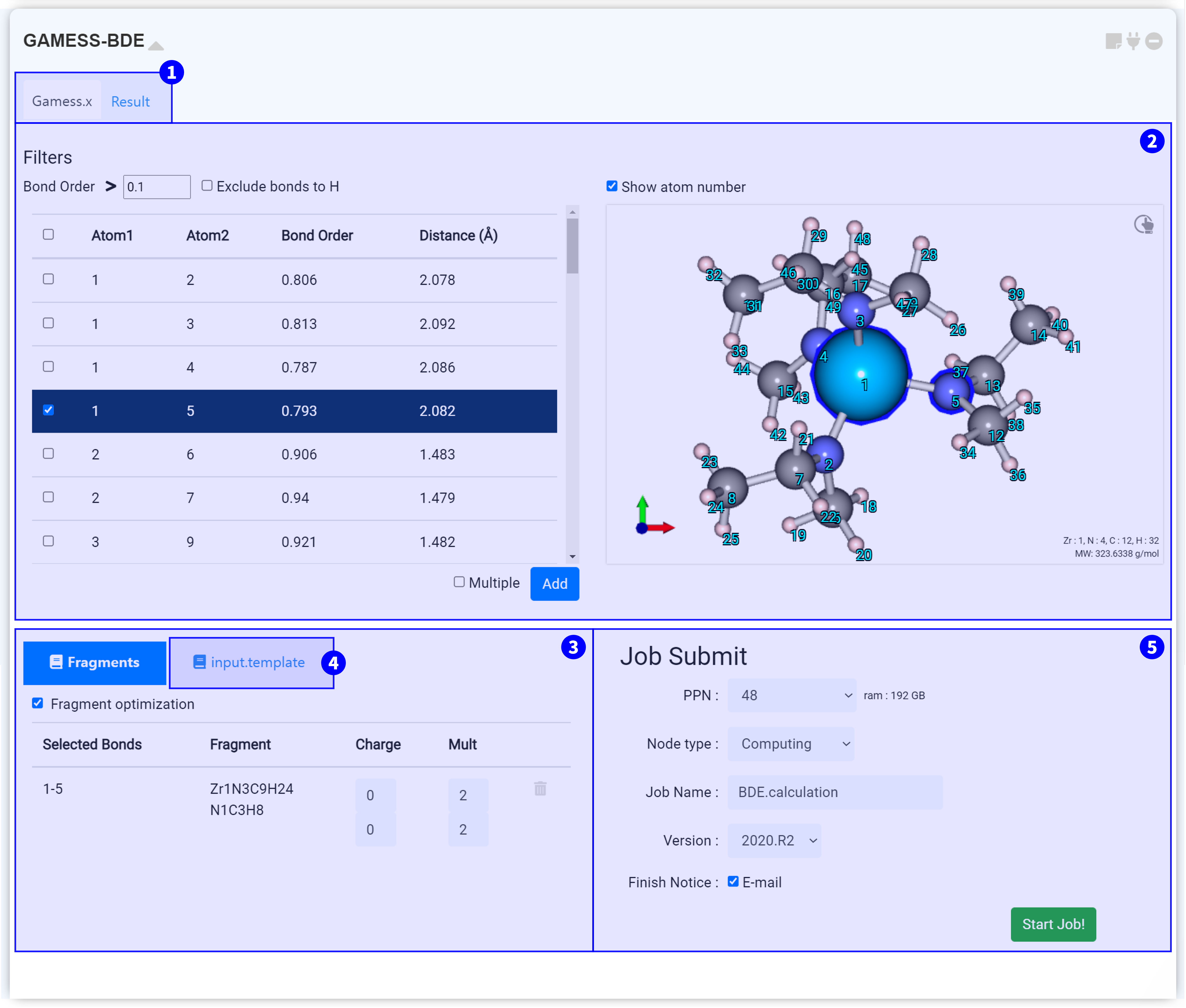
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS-BDE 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
원자간 결합 차수 및 거리를 확인할 수 있습니다. 리스트 상단 Filters의 ‘Bond order’ 값을 수정하여 원하는 값 이상의 결합 차수를 갖는 결합만을 표시하십시오. ‘Exclude bonds to H’ 체크박스를 통해 수소와의 결합을 표시할 것인지 여부를 결정할 수도 있습니다.
좌측 목록에서 결합을 선택하면, 우측 Visualizer에 선택한 결합에 참여하는 원자가 파란색 테두리로 표시됩니다. Visualizer 상단의 'Show atom number' 를 선택하여 원자의 고유 번호를 표시할 수 있습니다. Fragment를 만들 결합을 선택하고 버튼을 눌러 추가하십시오.
만일, 2 개 이상의 결합을 끊어 Fragment를 만드려면 'Multiple' 옵션을 선택하십시오. 이후 버튼을 눌러 생성될 Fragement를 색으로 확인할 수 있습니다.
생성한 Fragment 목록을 확인할 수 있습니다. 목록을 클릭하면 우측 상단 Visualizer에서 각 Fragment가 색으로 표시됩니다.
Fragment를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다.
①~④의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
재료과학 분야의 대표적인 예측 모델인 Crystal Graph Convolutional Neural Network (CGCNN)은 재료의 구조를 입력값으로 활용하여 재료의 물성을 결과값으로 예측하는 정방향 (direct) 예측 모델입니다. CGCNN 모듈은 CGCNN 모델을 사용하여 기계학습 계산을 수행할 수 있게 만든 모듈입니다. 이 모듈을 사용하면 사용자는 원하는 재료의 물성 (formation energy, band gap, bulk/shear moduli, poisson ratio 등)을 예측할 수 있습니다.
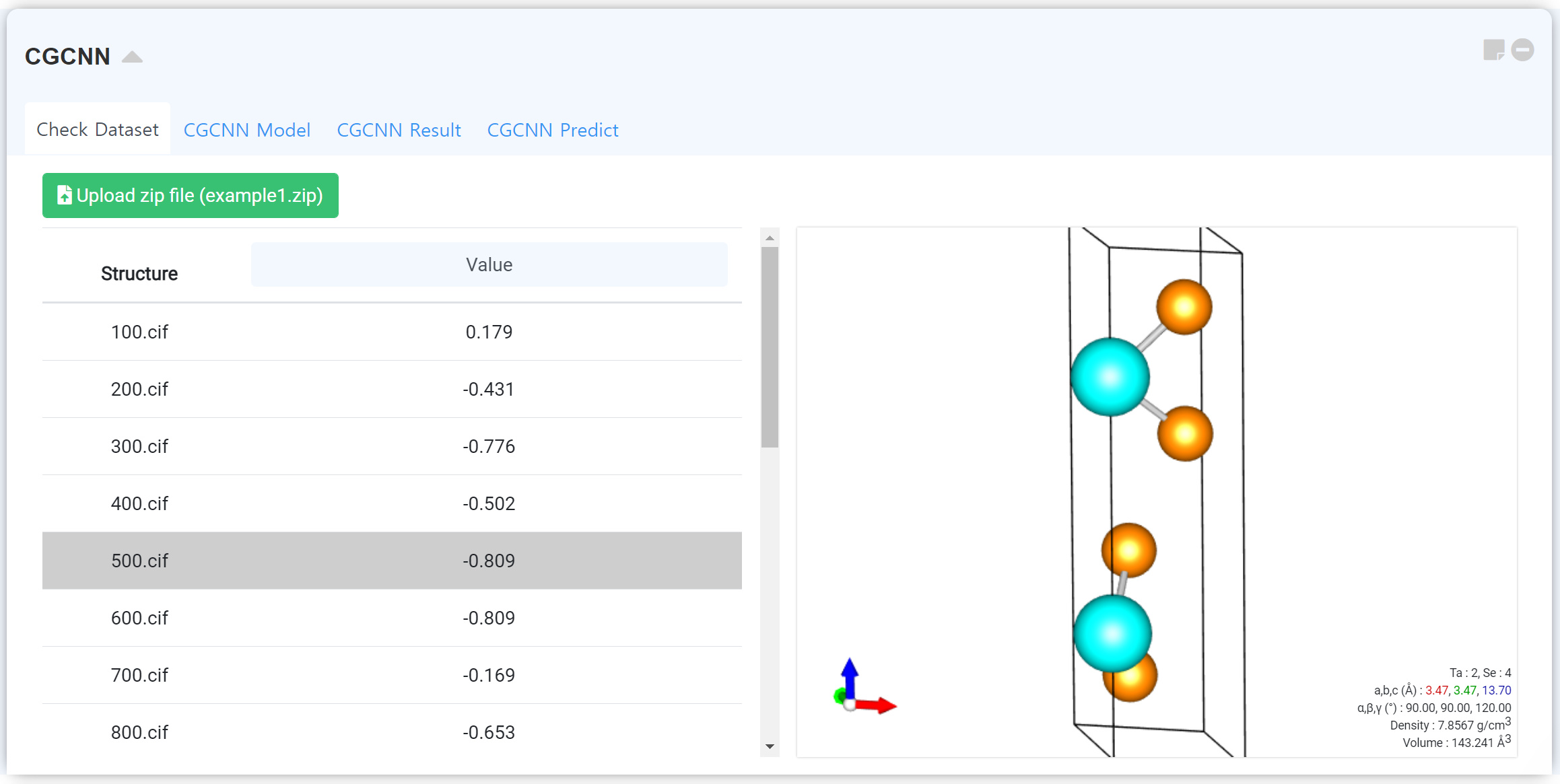
트레이닝 데이터셋을 업로드하고 확인할 수 있는 탭입니다. Upload zip file 버튼을 클릭하여 데이터베이스 압축 파일을 업로드하십시오. 데이터셋은 다음 예시를 참고하여 구성할 수 있습니다. 실제 학습 시에는 더 많은 수의 샘플이 필요합니다.
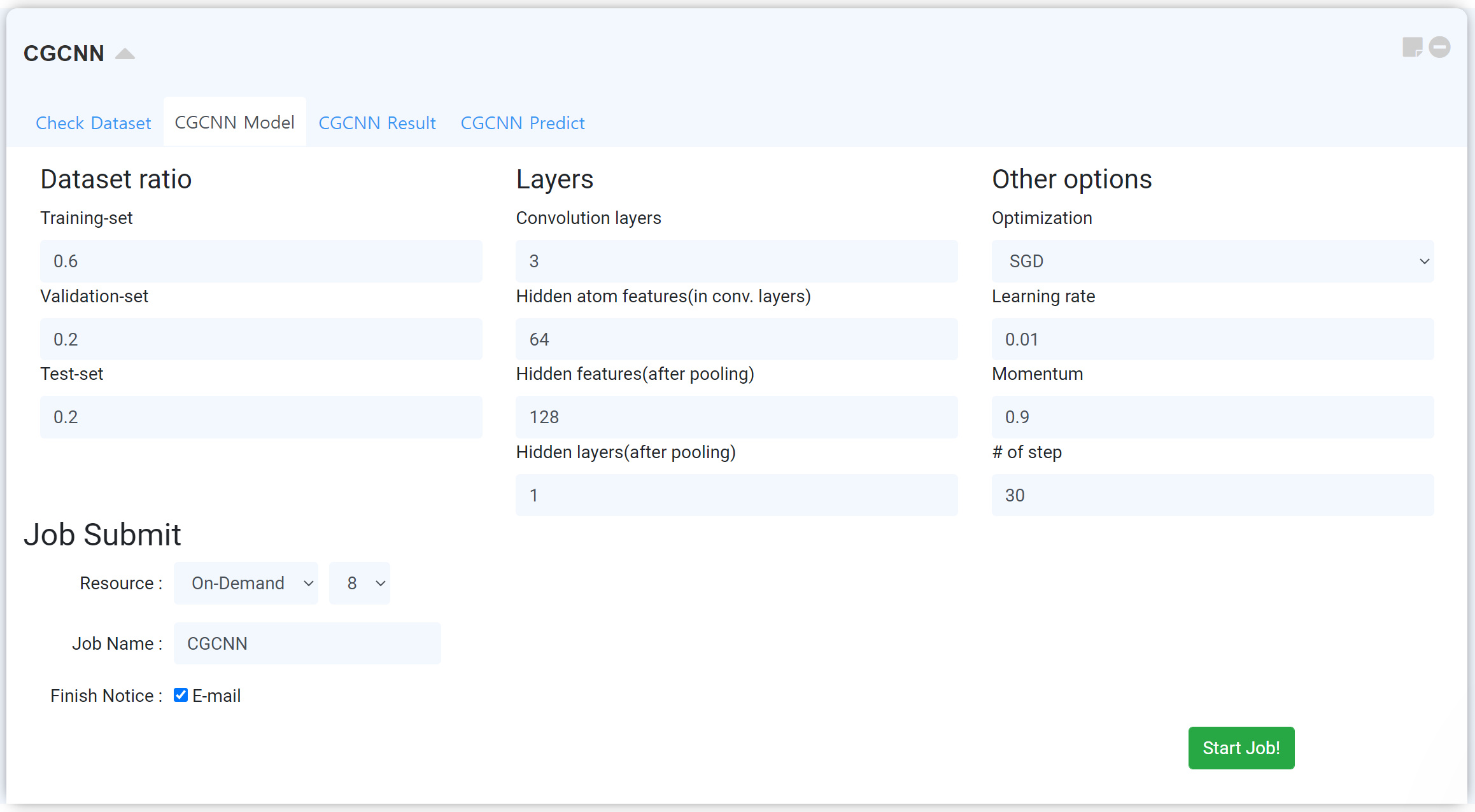
나만의 기계학습 모델을 만들 수 있는 영역입니다. 학습 데이터와 테스트 데이터를 어떻게 분배하여 기계 학습 모델을 만들 것인지, 모델을 만드는 데 필요한 하이퍼 파라미터를 어떻게 조절할 것인지, 그리고 학습에 어떠한 최적화 방법을 사용할 것인지 등을 결정하여 나만의 기계학습 모델을 만들 수 있습니다.
Dataset ratio 에서는 내가 가지고 있는 데이터 개수를 어떻게 나눠 활용할 것인지를 결정합니다. 만일 위 그림과 같이 설정할 경우, 데이터 중 약 60%는 Training-set에서 x,y의 상관관계를 찾는 학습에 사용하고, 나머지 20%를 Validation에 사용해 찾아낸 관계가 맞는지 확인합니다. 나머지 20%는 실전에 쓸 수 있도록 Test-set에 적용합니다.
이외에도 네트워크와 관련된 Layers, optimize와 관련된 Other options가 있습니다.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
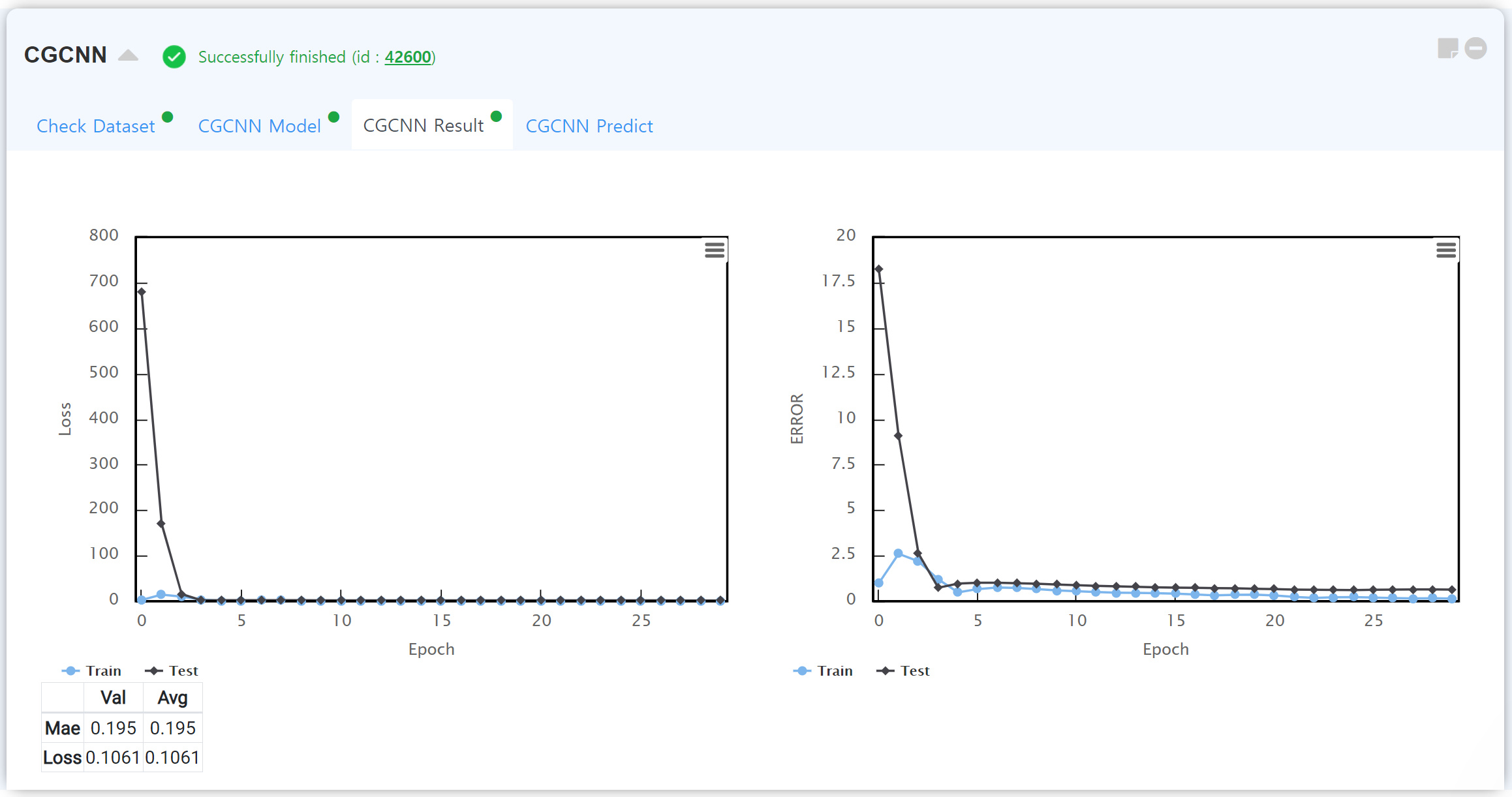
계산이 종료된 후, CGCNN로 얻은 학습 결과를 보여주는 탭입니다. 내가 설계한 기계학습 모델이 학습/테스트 데이터를 얼마나 정확히 예측하는지 학습 단계에 따른 Epoch를 확인할 수 있습니다. 일반적으로는 학습 데이터의 정확도가 테스트 데이터의 정확도보다 높기 때문에, 습득한 결과에서 학습 데이터의 정확도가 너무 높다면 과적합 (overfitting) 을 해결할 방법을 찾아야 합니다.
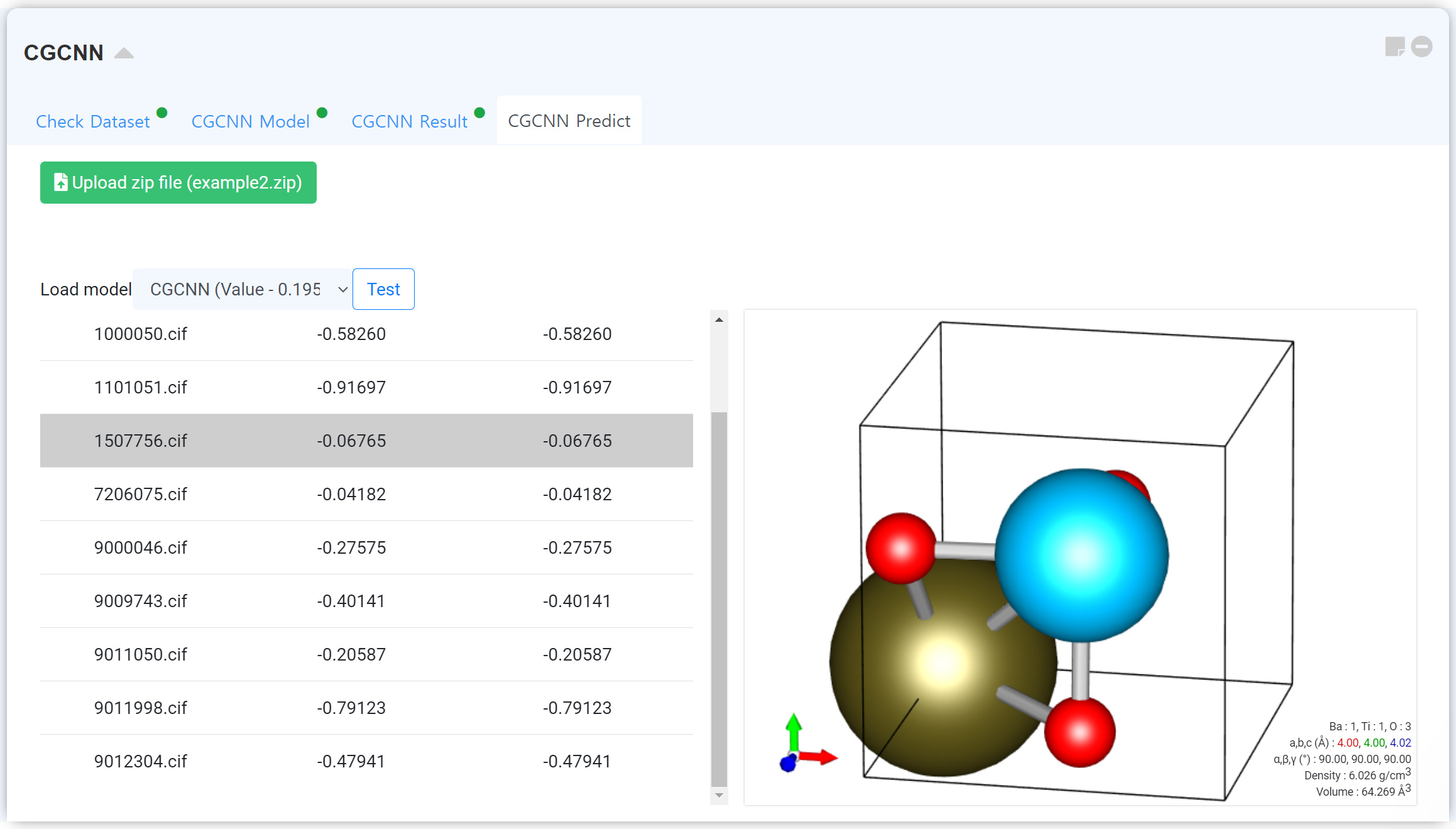
학습 종료 후, 계산된 모듈을 불러와서 업로드된 다른 구조의 물성을 예측할 수 있는 탭입니다. Upload zip file 버튼을 클릭하여 데이터베이스 압축 파일을 업로드하십시오. 데이터셋은 다음 예시를 참고하여 구성할 수 있습니다.
모듈 상단부에서는 두 개의 탭을 볼 수 있습니다. 'Gamess.x' 탭에서는 GAMESS-BDE 구동에 필요한 input script를 설정할 수 있습니다. 계산이 종료된 후, 'Result' 탭을 눌러 계산 결과를 확인할 수 있습니다.
원자간 결합 차수 및 거리를 확인할 수 있습니다. 리스트 상단 Filters의 ‘Bond order’ 값을 수정하여 원하는 값 이상의 결합 차수를 갖는 결합만을 표시하십시오. ‘Exclude bonds to H’ 체크박스를 통해 수소와의 결합을 표시할 것인지 여부를 결정할 수도 있습니다.
좌측 목록에서 결합을 선택하면, 우측 Visualizer에 선택한 결합에 참여하는 원자가 파란색 테두리로 표시됩니다. Visualizer 상단의 'Show atom number' 를 선택하여 원자의 고유 번호를 표시할 수 있습니다. Fragment를 만들 결합을 선택하고 버튼을 눌러 추가하십시오.
만일, 2 개 이상의 결합을 끊어 Fragment를 만드려면 'Multiple' 옵션을 선택하십시오. 이후 버튼을 눌러 생성될 Fragement를 색으로 확인할 수 있습니다.
생성한 Fragment 목록을 확인할 수 있습니다. 목록을 클릭하면 우측 상단 Visualizer에서 각 Fragment가 색으로 표시됩니다.
Fragment를 설정하면 input script가 자동으로 작성됩니다. Input script는 구조 정보와 keyword 설정 정보를 갖고 있으며 실제 서버에 전송되는 텍스트입니다. 따라서 설정한 값이 옳게 적용되었는지 항상 확인하는 것이 좋습니다.
①~④의 모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 (Job submit) 문서를 참고하여 계산 작업을 시작하십시오.
Materials Square는 LAMMPS를 쉽게 사용할 수 있도록 템플릿 모듈을 제공하고 있습니다. 템플릿 모듈의 모든 파라미터는 해당 계산에 최적화되어 있으므로, 단 몇 번의 클릭만으로 초기 조건을 설정하고 시뮬레이션을 수행할 수 있습니다. 또한 Custom 템플릿을 이용하면 Forcefield, Ensemble, Temperature, Simulation time만 조절하여 간단하게 원하는 MD 시뮬레이션을 수행할 수 있습니다.
- 원하는 데이터를 얻는 데 적합한 템플릿 모듈을 추가하십시오.
- 해당 시뮬레이션의 초기 조건을 설정하십시오. 자세한 설명은 다음 상세 설명을 확인하십시오.
- 작업 제출 (Job submit) 문서를 참고하여 작업을 시작하십시오.
- 시뮬레이션 종료 후 'Anaylsis' 탭에서 결과를 확인하십시오. 자세한 설명은 다음 '템플릿 상세 설명' 을 참고하십시오.
- Thermalization
- Tg/CTE
- Elastic properties
- Dielectric constant
- Solubility parameter
- Viscosity (EMD)
- Viscosity (NEMD)
'Cascade' 모듈은 조사 손상 시뮬레이션을 수행하기 위한 전용 LAMMPS 모듈입니다. 'Cascade simulation'은 충돌 캐스케이드 (collision cascade)라고도 하며, 조사된 고에너지 원자 (PKA)에 의한 구조 변화를 모사합니다.
Cascade simulation에는 매우 큰 모델이 필요하므로, Structure builder에서 직접 모델링하는 대신 LAMMPS (CAS) 모듈에서 시뮬레이션 모델 사이즈를 결정합니다. 그러므로, LAMMPS (CAS) 모듈을 통해 cascade simulation을 수행할 때는 unit cell을 모델링한 structure builder 모듈에 연결하십시오.
Primary Knock-on Atom (PKA)는 (0, 0, 0) 위치의 원자로 자동 선택됩니다.
Cascade simulation은 다음과 같은 단계로 진행됩니다.
설정한 총 시뮬레이션 시간은 반으로 나누어 Cascade와 Relaxation 단계에 각각 배분됩니다. 그러나 timestep이 다르므로 같은 시간 당 step의 수는 달라집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
LAMMPS (CAS) 모듈의 Analysis 탭은 Movie 모듈과 그래프 모듈로 이루어져 있습니다. Movie에서 trajectory를 확인하고, 하단 그래프에서 시간에 따라 변화하는 defect의 수를 확인할 수 있습니다.
Cascade simulation에는 매우 큰 모델이 필요하므로, Structure builder에서 직접 모델링하는 대신 LAMMPS (CAS) 모듈에서 시뮬레이션 모델 사이즈를 결정합니다. 그러므로, LAMMPS (CAS) 모듈을 통해 cascade simulation을 수행할 때는 unit cell을 모델링한 structure builder 모듈에 연결하십시오.
Primary Knock-on Atom (PKA)는 (0, 0, 0) 위치의 원자로 자동 선택됩니다.
- 해당 시스템에 가장 적합한 Forcefield를 선택하십시오.
- 시뮬레이션 모델의 크기를 결정하십시오. 시스템이 작을 경우 주기경계조건에 의해 PKA가 처음 위치로 돌아올 수 있습니다. 또한, 시스템이 커지면 계산 시간이 증가하는 것에 유의하십시오.
- PKA Direction을 설정하십시오.
를 누르면 무작위로 벡터를 결정할 수 있습니다. - PKA Energy를 설정하십시오.
PKA Energy는 cascade simulation 시 PKA의 초기 속도를 결정합니다. - Thermailization 시의 온도를 결정하십시오.
- 총 시뮬레이션 시간을 결정하십시오.
Cascade simulation은 다음과 같은 단계로 진행됩니다.
- Relaxation
- Thermalization
- Cascade : NVE 1
(Velocity 설정, timestep 0.01 fs) - Relaxation : NVE 2
(timestep 0.03 fs)
설정한 총 시뮬레이션 시간은 반으로 나누어 Cascade와 Relaxation 단계에 각각 배분됩니다. 그러나 timestep이 다르므로 같은 시간 당 step의 수는 달라집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
LAMMPS (CAS) 모듈의 Analysis 탭은 Movie 모듈과 그래프 모듈로 이루어져 있습니다. Movie에서 trajectory를 확인하고, 하단 그래프에서 시간에 따라 변화하는 defect의 수를 확인할 수 있습니다.
Equation of State (EOS)를 계산하기 위한 전용 LAMMPS 모듈입니다. 모듈에서의 간단한 조작을 통해 해당 시스템의 기본적인 기하 성질 (geometric property), 즉 바닥 상태에서의 부피 및 그 에너지, 그리고 'bulk modulus' 를 얻을 수 있습니다. Unit cell이 모델링된 Structure builder 모듈에 연결하여 사용하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
(E0: 가장 안정한 부피에서의 에너지, B0: Bulk modulus, Bp: Bulk modulus의 미분값, V0: 가장 안정한 부피)
- 해당 시스템에 대해 가장 적절한 Potential을 선택하십시오.
- EOS를 그리기 위해 변화시킬 부피의 최대 변경비율을 결정하십시오.
- 부피가 변경될 간격을 결정하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
(E0: 가장 안정한 부피에서의 에너지, B0: Bulk modulus, Bp: Bulk modulus의 미분값, V0: 가장 안정한 부피)
열전도도 (Thermal conductivity)를 계산하기 위한 전용 LAMMPS 템플릿 모듈입니다. NEMD (Non-equilibrium molucular dynamics) 방식을 통해 격자 열전도도 계산을 수행합니다. '열전도도 (Thermal conductivity)', '온도 분포 (Temperature distribution)', '열유속 (Heat flux)'과 같은 열 특성을 얻을 수 있습니다.
열전달 시뮬레이션을 위해서는 충분히 큰 모델을 사용해야 합니다. 이때 Structure builder에서 직접 모델링하는 대신 LAMMPS 모듈의 'Supercell' 옵션을 통해 시뮬레이션 모델 사이즈를 결정합니다. 따라서 해당 계산을 수행하기 위하여, LAMMPS 모듈을 직육면체 단위 셀 (orthogonal)을 모델링한 'structure builder' 모듈에 연결하십시오.
시뮬레이션에 사용되는 모델은 다음과 같은 형태를 갖게 됩니다.
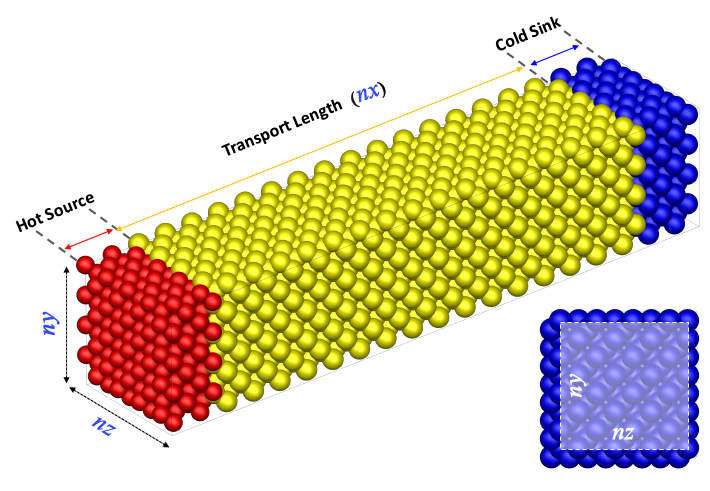
모델은 열이 가해지는 'Hot Source', 열 전달이 발생하는 'Transport Length', 저온 영역인 'Cold Sink' 영역으로 나눌 수 있습니다. 모델의 양 끝단에 위치한 단위 셀은 고정하여 실험에서의 suspended 영역으로 간주하며, 고정 영역 안쪽 20 unit cell에 각각 다른 온도를 적용하여 'Hot Source', 'Cold Sink' 영역으로 지정합니다. 'Transport Length' 는 총 길이로부터 42 unit cell 길이를 제한 나머지 부분입니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
LAMMPS (Thermal Conductivity) 모듈의 'Analysis' 탭은 결과 데이터 및 세 개의 그래프로 이루어져 있습니다.
얻어진 열전도도 값과 모델의 길이, 단면적이 표시됩니다.
시뮬레이션의 진행에 따라 달라지는 각 영역의 온도 분포를 나타냅니다.
단위 부피·단위 시간에 대한 열유량 (heat flux)의 변화를 나타냅니다.
축적되는 에너지를 시간에 따라 나타냅니다.
하단의 'Slope' 를 조절하면 추세선의 기울기를 변경하여 새로 계산된 'Thermal Conductivity' 값을 확인할 수 있습니다.
열전달 시뮬레이션을 위해서는 충분히 큰 모델을 사용해야 합니다. 이때 Structure builder에서 직접 모델링하는 대신 LAMMPS 모듈의 'Supercell' 옵션을 통해 시뮬레이션 모델 사이즈를 결정합니다. 따라서 해당 계산을 수행하기 위하여, LAMMPS 모듈을 직육면체 단위 셀 (orthogonal)을 모델링한 'structure builder' 모듈에 연결하십시오.
시뮬레이션에 사용되는 모델은 다음과 같은 형태를 갖게 됩니다.
모델은 열이 가해지는 'Hot Source', 열 전달이 발생하는 'Transport Length', 저온 영역인 'Cold Sink' 영역으로 나눌 수 있습니다. 모델의 양 끝단에 위치한 단위 셀은 고정하여 실험에서의 suspended 영역으로 간주하며, 고정 영역 안쪽 20 unit cell에 각각 다른 온도를 적용하여 'Hot Source', 'Cold Sink' 영역으로 지정합니다. 'Transport Length' 는 총 길이로부터 42 unit cell 길이를 제한 나머지 부분입니다.
- 해당 시스템에 가장 적합한 potential을 선택하십시오.
- 열 전달이 발생할 축의 방향을 지정하십시오.
- 시뮬레이션 모델의 크기를 결정하십시오.
- 중심 영역의 온도를 결정하십시오. (ΔT는 30 K로 고정)
- 시뮬레이션 시간을 설정하십시오.
- 시뮬레이션의 각 시간 단계 사이의 간격을 지정하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
LAMMPS (Thermal Conductivity) 모듈의 'Analysis' 탭은 결과 데이터 및 세 개의 그래프로 이루어져 있습니다.
얻어진 열전도도 값과 모델의 길이, 단면적이 표시됩니다.
시뮬레이션의 진행에 따라 달라지는 각 영역의 온도 분포를 나타냅니다.
단위 부피·단위 시간에 대한 열유량 (heat flux)의 변화를 나타냅니다.
축적되는 에너지를 시간에 따라 나타냅니다.
하단의 'Slope' 를 조절하면 추세선의 기울기를 변경하여 새로 계산된 'Thermal Conductivity' 값을 확인할 수 있습니다.
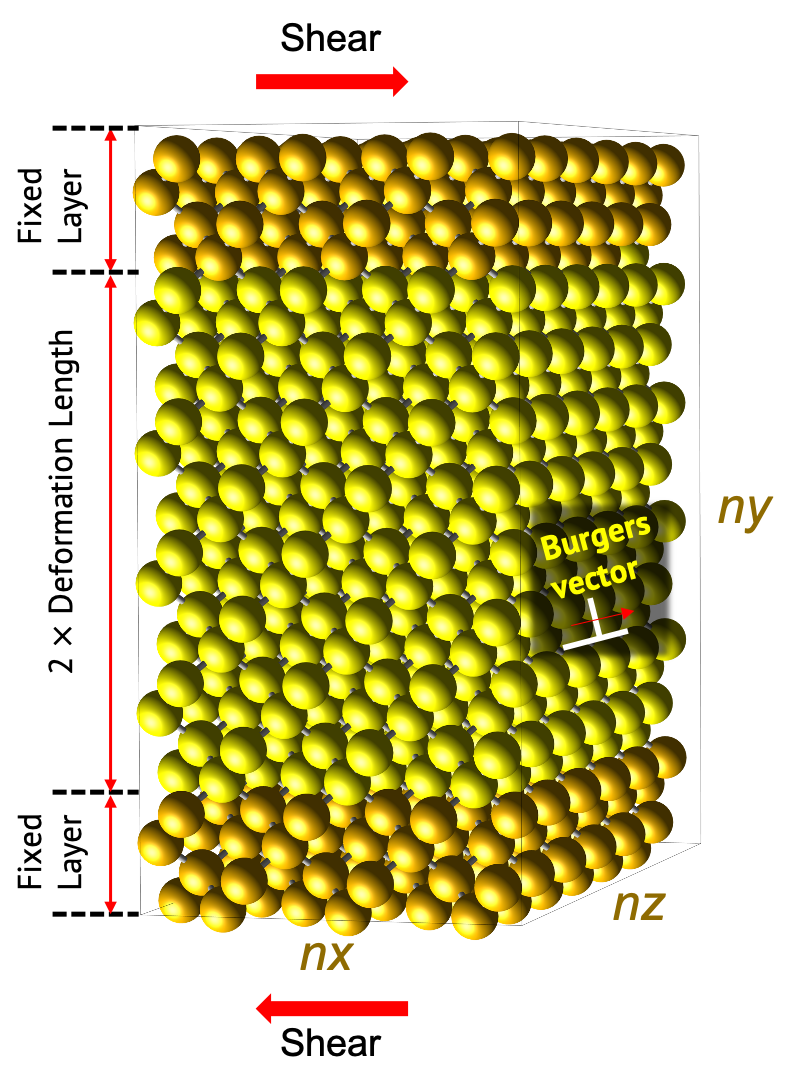
Dislocation의 움직임과 재료의 소성 특성을 평가하기 위한 전용 LAMMPS 템플릿 모듈입니다. 'Dislocation' 모듈을 통해 임계 분해 전단 응력 (critical resolved shear stress)을 계산하여 전위 (dislocation)의 이동성을 평가할 수 있습니다. 전단 변형 (shear deformation) 동안의 응력-변형률 곡선 (stress-strain curve)과 구조 변화 (structure variations) 정보를 얻을 수 있습니다.
Dislocation simulation을 위해서는 충분히 큰 모델을 사용해야 합니다. 이때 Structure builder에서 직접 모델링하는 대신 LAMMPS 모듈의 'Supercell' 옵션을 통해 시뮬레이션 모델의 크기를 결정합니다. 따라서 해당 계산을 수행하기 위하여, LAMMPS 모듈을 직육면체 단위 셀 (orthogonal)을 모델링한 structure builder 모듈에 연결하십시오.
시뮬레이션에 사용되는 모델은 다음과 같은 형태를 갖게 됩니다.
모델은 edge dislocation과 screw dislocation 중 하나를 선택할 수 있습니다. 두 모델은 공통적으로 힘이 가해지는 Fixed layer와 변형이 일어나는 Deformation region으로 나눌 수 있습니다. Fixed layer는 고정하여 실험에서의 suspended 영역으로 간주하며, 해당 unitcell에서 최인접 거리의 10배에 해당되는 길이로 설정되어 있습니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다. LAMMPS (Dislocation) 모듈의 'Analysis' 탭은 결과 데이터 및 그래프로 이루어져 있습니다.
샘플의 구조를 분석합니다.
Dislocation의 burger's vector와 line 길이를 분석해 줍니다.
전단 변형률에 다른 전단 응력을 나타냅니다.
Dislocation simulation을 위해서는 충분히 큰 모델을 사용해야 합니다. 이때 Structure builder에서 직접 모델링하는 대신 LAMMPS 모듈의 'Supercell' 옵션을 통해 시뮬레이션 모델의 크기를 결정합니다. 따라서 해당 계산을 수행하기 위하여, LAMMPS 모듈을 직육면체 단위 셀 (orthogonal)을 모델링한 structure builder 모듈에 연결하십시오.
시뮬레이션에 사용되는 모델은 다음과 같은 형태를 갖게 됩니다.
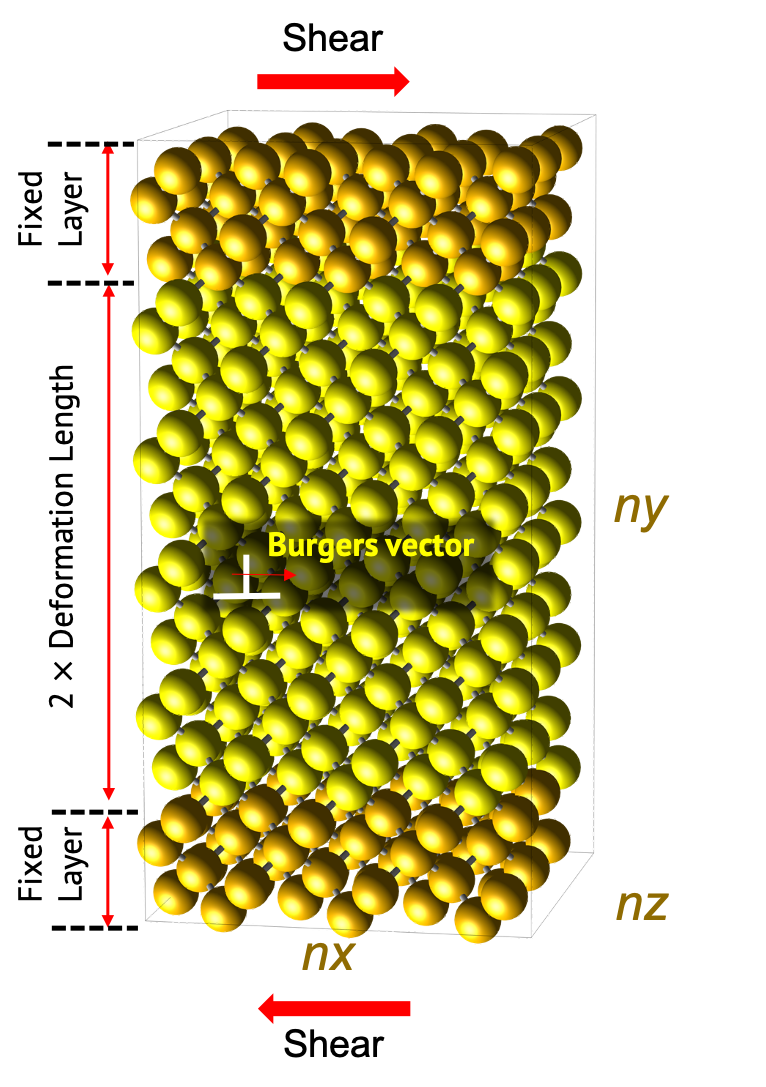 |  |
모델은 edge dislocation과 screw dislocation 중 하나를 선택할 수 있습니다. 두 모델은 공통적으로 힘이 가해지는 Fixed layer와 변형이 일어나는 Deformation region으로 나눌 수 있습니다. Fixed layer는 고정하여 실험에서의 suspended 영역으로 간주하며, 해당 unitcell에서 최인접 거리의 10배에 해당되는 길이로 설정되어 있습니다.
-
FCC <110>{111} slip system : →
 → FCC (111) 선택
→ FCC (111) 선택
-
BCC <111>{110} slip system : → → BCC (110) 선택
- 해당 시스템에 가장 적합한 Forcefield를 선택하십시오.
- 구조의 계열을 선택하십시오. (FCC or BCC)
- 시뮬레이션 모델의 크기를 결정하십시오.
- 원하는 dislocation을 선택하십시오. (Edge or Screw)
- 'Poisson's ratio' 를 설정하십시오. (금속은 보통 0.3)
- Shear 방향으로 displacement를 결정하십시오.
- 총 전단 변형률 (total shear strain) 을 지정하십시오.
- Dislocation normal 방향과 glide 방향은 적당히 길어야 보다 정확한 결과를 얻을 수 있습니다.
- Edge 시뮬레이션의 경우, Z 방향으로는 supercell을 최소 3 이상 복제하도록 설정하는 것을 추천합니다.
- Screw 시뮬레이션의 경우, shear 방향인 X 방향과 glide 방향인 Z 방향 모두 적당히 길어야 합니다.
- Supercell이 너무 크면 많은 양의 크레딧이 과금될 수 있습니다.
- Displacement를 너무 크게 설정하면 계산 시간은 빠르나 계산이 부정확할 수 있으며, 반면 너무 작으면 오랜 시간이 걸리게 됩니다.
- Total shear strain 이 작으면 elastic 영역만 계산하게 됩니다. 따라서 적당한 total shear strain을 입력해야 합니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다. LAMMPS (Dislocation) 모듈의 'Analysis' 탭은 결과 데이터 및 그래프로 이루어져 있습니다.
샘플의 구조를 분석합니다.
Dislocation의 burger's vector와 line 길이를 분석해 줍니다.
전단 변형률에 다른 전단 응력을 나타냅니다.
인장 시험 (Tensile test) 시뮬레이션을 위한 LAMMPS 모듈입니다. 모듈에서의 간단한 조작을 통해 해당 시스템의 인장 강도 (tensile strength)를 알아볼 수 있습니다. 해당 템플릿을 선택하고, Unit cell이 모델링된 Structure builder 모듈에 연결하여 사용하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
영률 (Young' Modulus)과 극한 인장 강도 (Ultimate Tensile Strength (GPa)) 결과 및 Trajectory movie, Stress-strain curve가 표시됩니다. 영률과 극한 인장 강도는 일반적인 힘-인장 곡선에서의 예측값이므로 실제 값과 차이가 있을 수 있습니다.
- 해당 시스템에 대해 가장 적절한 Potential을 선택하십시오.
- 인장 강도를 측정할 방향을 선택하십시오.
- 시뮬레이션 모델의 크기를 설정하십시오. 연결한 Structure builder의 모델을 입력 숫자만큼 복제하여 supercell을 생성합니다.
- 시뮬레이션 온도를 지정하십시오.
- 구조 변형을 어느 정도로 가할 것인지 결정하십시오. (1. 일정한 변형률 (ps-1), 2. 일정한 속도 (Å/ps))
N.B. 총 인장율은 'Strain rate (ps-1) * Time (ps)' 로 계산할 수 있습니다. - 총 시뮬레이션 시간을 결정하십시오.
- 시간 적분을 수행할 시간 간격을 결정하십시오. (timestep)
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
영률 (Young' Modulus)과 극한 인장 강도 (Ultimate Tensile Strength (GPa)) 결과 및 Trajectory movie, Stress-strain curve가 표시됩니다. 영률과 극한 인장 강도는 일반적인 힘-인장 곡선에서의 예측값이므로 실제 값과 차이가 있을 수 있습니다.
"Melting & Quenching" 모듈은 시스템의 온도를 급격히 증가 (용융, Melting)시키거나 또는 감소 (급랭, Quenching) 시킬 수 있습니다. 일반적으로, 이 모듈은 주어진 온도에서 비정질 구조 생성에 사용할 수 있으며 방사형 분포 함수 (RDF), 각도 분포 함수 (ADF) 및 온도 프로파일이 제공됩니다. 해당 템플릿을 선택하고, orthogonal cell이 모델링된 structure builder 모듈에 연결하여 사용하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Trajectory movie, 방사형 분포 함수 (RDF), 각도 분포 함수 (ADF) 및 온도 프로파일을 확인할 수 있습니다.
- 해당 시스템에 대해 가장 적절한 Potential을 선택하십시오.
- 시뮬레이션 모델의 크기를 설정하십시오. 연결한 Structure builder의 모델을 입력 숫자만큼 복제하여 supercell을 생성합니다.
- 시뮬레이션 시작, 최종 온도를 지정하십시오.
- Quenching (melting) rate을 결정하십시오.
- 시스템에 가해질 압력을 결정하십시오.
- Number of bins를 설정하여 cutoff radius를 몇 개의 구간으로 자를 지 결정하십시오.
- 총 시뮬레이션 시간을 결정하십시오.
- 시간 적분을 수행할 시간 간격을 결정하십시오. (timestep)
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Trajectory movie, 방사형 분포 함수 (RDF), 각도 분포 함수 (ADF) 및 온도 프로파일을 확인할 수 있습니다.
LAMMPS (Custom) 모듈은 템플릿으로 제공되지 않는 사용자 정의 시뮬레이션을 수행할 수 있도록 준비된 모듈입니다. 알맞은 forcefield를 선택하고, 초기 조건을 지정하십시오.
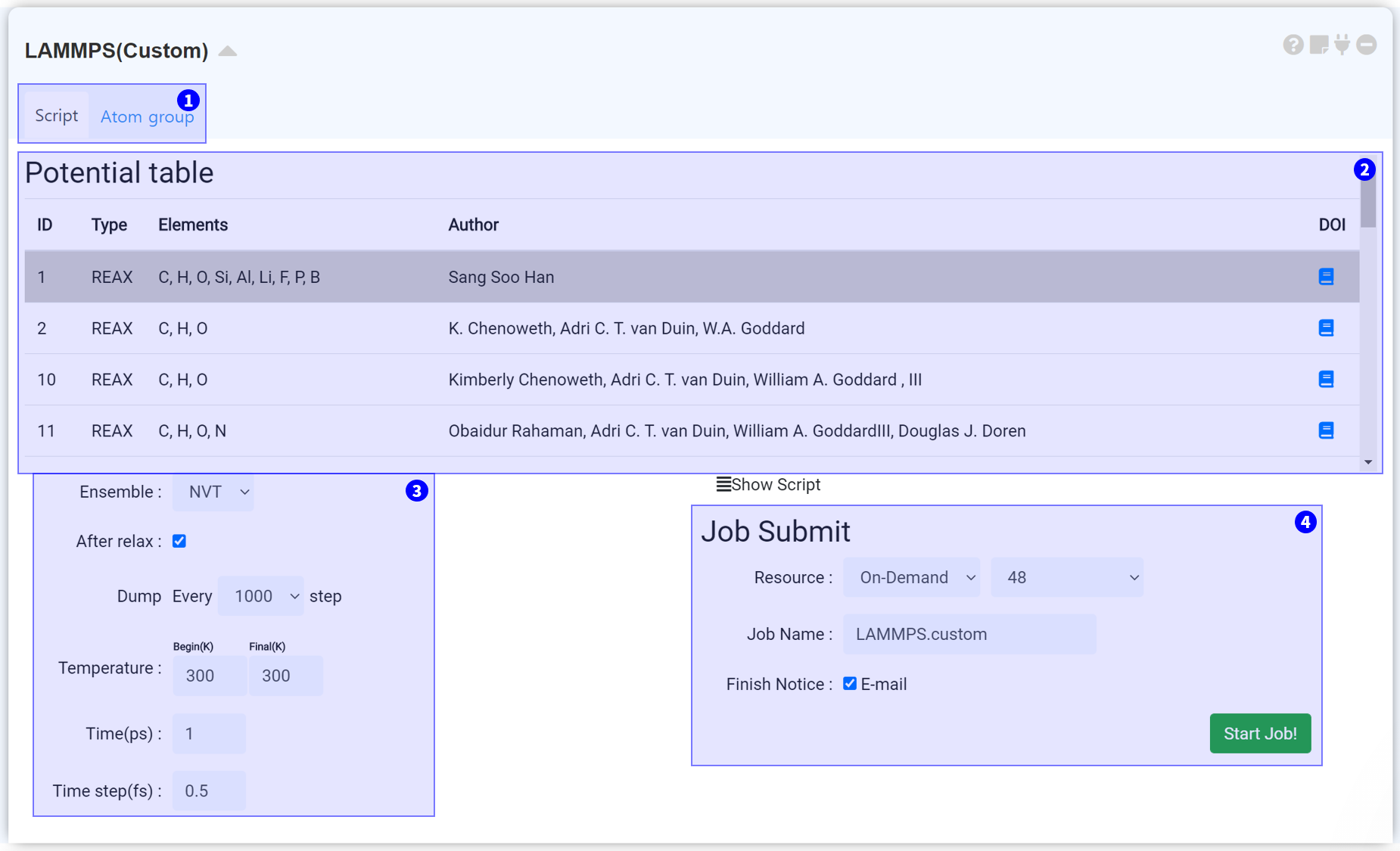
LAMMPS (Custom) 모듈은 크게 두 가지 탭으로 구성되어 있습니다. Script 탭에서는 Potential과 Input parameters를 결정할 수 있으며, 설정한 데이터를 서버에 전송하여 시뮬레이션 작업을 시작할 수 있습니다. Atom group 탭은 특정 원자 그룹을 설정하여 시뮬레이션을 정밀하게 조정하기 위한 탭으로, 새 'atom group' 을 만든 후 속력, 힘, 온도 등을 조절하십시오. 자세한 내용은 아래 'Grouping' 섹션을 참고하십시오.
Molecular Dynamics 시뮬레이션에 필수적인 Potential을 선택할 수 있는 리스트입니다. 현재 Reactive Forcefield를 기본적으로 제공하고 있으며, 추후 다른 MD potential을 추가할 수 있도록 지원할 예정입니다.
MD 시뮬레이션을 위한 LAMMPS의 input parameter를 설정할 수 있습니다. 현재 Ensemble, relax 여부, Dump, Temperature, Time을 조절할 수 있습니다.
LAMMPS input 설정에 대한 자세한 정보는 appendix를 참고하십시오.
모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 문서를 참조하여 작업을 제출하십시오.
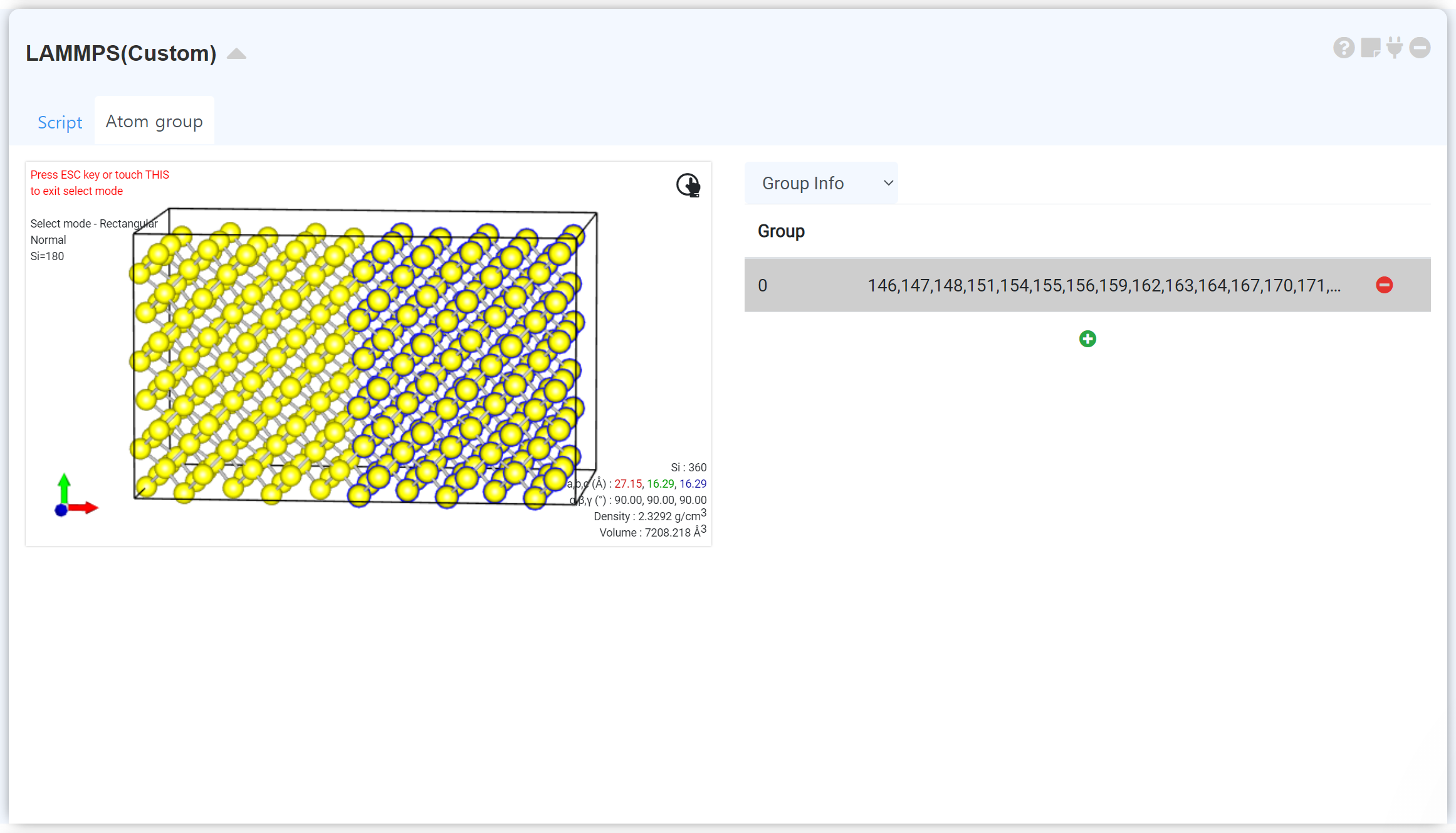
LAMMPS (Custom) 에서는 일부 원자를 그룹으로 설정하여 초기 조건을 부여할 수 있습니다. LAMMPS 모듈의 'Atom Group' 탭을 선택하면, 모듈을 Structure builder에 연결한 시점에서의 구조 정보가 담긴 visualizer가 표시됩니다. 우클릭 팝업에서 Select 메뉴를 활성화한 다음 추가적인 설정을 원하는 원자들을 선택하십시오. +를 누르면 선택한 원자들에 대해 Atom group이 생성됩니다.
Atom Group 추가에는 개수 제한이 없으며, 리스트에서 기존에 지정한 Atom group을 클릭하면 해당 Atom group에 속하는 원자들을 선택하여 보여주므로 편리합니다.
초기 조건 설정을 원하는 그룹을 선택하고 Group Info를 클릭한 다음 원하는 조건을 선택하고 체크박스에 체크한 다음 각 축 방향으로 가해질 조건을 설정하십시오.
LAMMPS (Custom) 모듈은 크게 두 가지 탭으로 구성되어 있습니다. Script 탭에서는 Potential과 Input parameters를 결정할 수 있으며, 설정한 데이터를 서버에 전송하여 시뮬레이션 작업을 시작할 수 있습니다. Atom group 탭은 특정 원자 그룹을 설정하여 시뮬레이션을 정밀하게 조정하기 위한 탭으로, 새 'atom group' 을 만든 후 속력, 힘, 온도 등을 조절하십시오. 자세한 내용은 아래 'Grouping' 섹션을 참고하십시오.
Molecular Dynamics 시뮬레이션에 필수적인 Potential을 선택할 수 있는 리스트입니다. 현재 Reactive Forcefield를 기본적으로 제공하고 있으며, 추후 다른 MD potential을 추가할 수 있도록 지원할 예정입니다.
MD 시뮬레이션을 위한 LAMMPS의 input parameter를 설정할 수 있습니다. 현재 Ensemble, relax 여부, Dump, Temperature, Time을 조절할 수 있습니다.
LAMMPS input 설정에 대한 자세한 정보는 appendix를 참고하십시오.
모든 설정을 완료했다면 작업을 제출할 준비가 된 것입니다. 작업 제출 문서를 참조하여 작업을 제출하십시오.
LAMMPS (Custom) 에서는 일부 원자를 그룹으로 설정하여 초기 조건을 부여할 수 있습니다. LAMMPS 모듈의 'Atom Group' 탭을 선택하면, 모듈을 Structure builder에 연결한 시점에서의 구조 정보가 담긴 visualizer가 표시됩니다. 우클릭 팝업에서 Select 메뉴를 활성화한 다음 추가적인 설정을 원하는 원자들을 선택하십시오. +를 누르면 선택한 원자들에 대해 Atom group이 생성됩니다.
Atom Group 추가에는 개수 제한이 없으며, 리스트에서 기존에 지정한 Atom group을 클릭하면 해당 Atom group에 속하는 원자들을 선택하여 보여주므로 편리합니다.
초기 조건 설정을 원하는 그룹을 선택하고 Group Info를 클릭한 다음 원하는 조건을 선택하고 체크박스에 체크한 다음 각 축 방향으로 가해질 조건을 설정하십시오.
특정 온도 및 normal density에서 열적평형을 이루는 densely packed simulated polymer 모델을 만들기 위한 모듈입니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Trajectory movie, Temperature, Volume, Energy를 볼 수 있습니다.
- 해당 시스템에 대해 가장 적절한 Potential을 선택하십시오. 'Dreiding' 이 기본으로 선택되어 있습니다.
- 기본 값 (Bond scale, Charge equilibrate method, Long range interaction distance, Coulombic interaction) 을 확인하십시오.
- 을 눌러 구조 사전 최적화를 진행하십시오. 원자 개수가 많은 경우 버튼을 눌러 최적화 작업을 클라우드 서버에 제출하는 것이 좋습니다. (분자 구조 복잡도에 따라 재현이 어려울수 있습니다.)
- 시스템의 온도를 결정하십시오.
- 시스템이 최종적으로 가지게 될 밀도를 지정하십시오.
- 필요한 경우, Advanced Options를 열어 계산 조건을 보다 상세히 설정하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Trajectory movie, Temperature, Volume, Energy를 볼 수 있습니다.
고분자 구조의 유리전이온도 (Glass transition temperature) 및 열팽창계수 (Constant of thermal expansion, CTE)를 계산하기 위한 모듈입니다.
LAMMPS (Tg/CTE) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Trajectory movie, Volume vs Temperature graph, CTE vs Volume, Temperature change, MSD 데이터를 볼 수 있습니다.
LAMMPS (Tg/CTE) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
- 계산의 정확도를 설정하십시오. 이는 데이터의 개수에 영향을 미칩니다. 다시 말해, 데이터 사이의 온도 간격을 결정합니다.
- 유리전이 시뮬레이션을 수행할 시작 온도 및 종료 온도를 결정하십시오.
- 필요한 경우, Advanced Options를 열어 계산 조건을 보다 상세히 설정하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Trajectory movie, Volume vs Temperature graph, CTE vs Volume, Temperature change, MSD 데이터를 볼 수 있습니다.
고분자 구조의 탄성 특성을 계산하기 위한 모듈입니다.
LAMMPS (Elastic properties) 모듈은 유리전이온도 계산에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Tg/CTE) 에 연결하십시오.
Forcefield 등의 정보는 연결된 LAMMPS (Tg/CTE) 모듈로부터 가져와집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Bulk modulus, Shear modulus, Young's modulus, Poisson's ratio 데이터를 볼 수 있습니다.
LAMMPS (Elastic properties) 모듈은 유리전이온도 계산에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Tg/CTE) 에 연결하십시오.
Forcefield 등의 정보는 연결된 LAMMPS (Tg/CTE) 모듈로부터 가져와집니다.
- 계산의 정확도 및 시뮬레이션 시작 온도 및 종료 온도를 확인하십시오.
- 필요한 경우, Advanced Options를 열어 time step을 조절하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
Bulk modulus, Shear modulus, Young's modulus, Poisson's ratio 데이터를 볼 수 있습니다.
Dielectric constant 계산을 통해 고분자 구조의 전기적 특성을 알아볼 수 있습니다.
LAMMPS (Dielectric constant) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
이때, Coulombic interaction 옵션이 활성화된 thermalization 계산을 사용하는 것이 좋습니다.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
온도에 따라 변화하는 유전 상수 데이터를 볼 수 있습니다.
LAMMPS (Dielectric constant) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
이때, Coulombic interaction 옵션이 활성화된 thermalization 계산을 사용하는 것이 좋습니다.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
- 계산의 정확도를 설정하십시오. 이는 데이터의 개수에 영향을 미칩니다. 다시 말해, 데이터 사이의 온도 간격을 결정합니다.
- 시뮬레이션을 수행할 시작 온도 및 종료 온도를 결정하십시오.
- 필요한 경우, Advanced Options를 열어 계산 조건을 보다 상세히 설정하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
온도에 따라 변화하는 유전 상수 데이터를 볼 수 있습니다.
고분자의 Hildebrand Solubility Parameter를 계산하기 위한 모듈입니다.
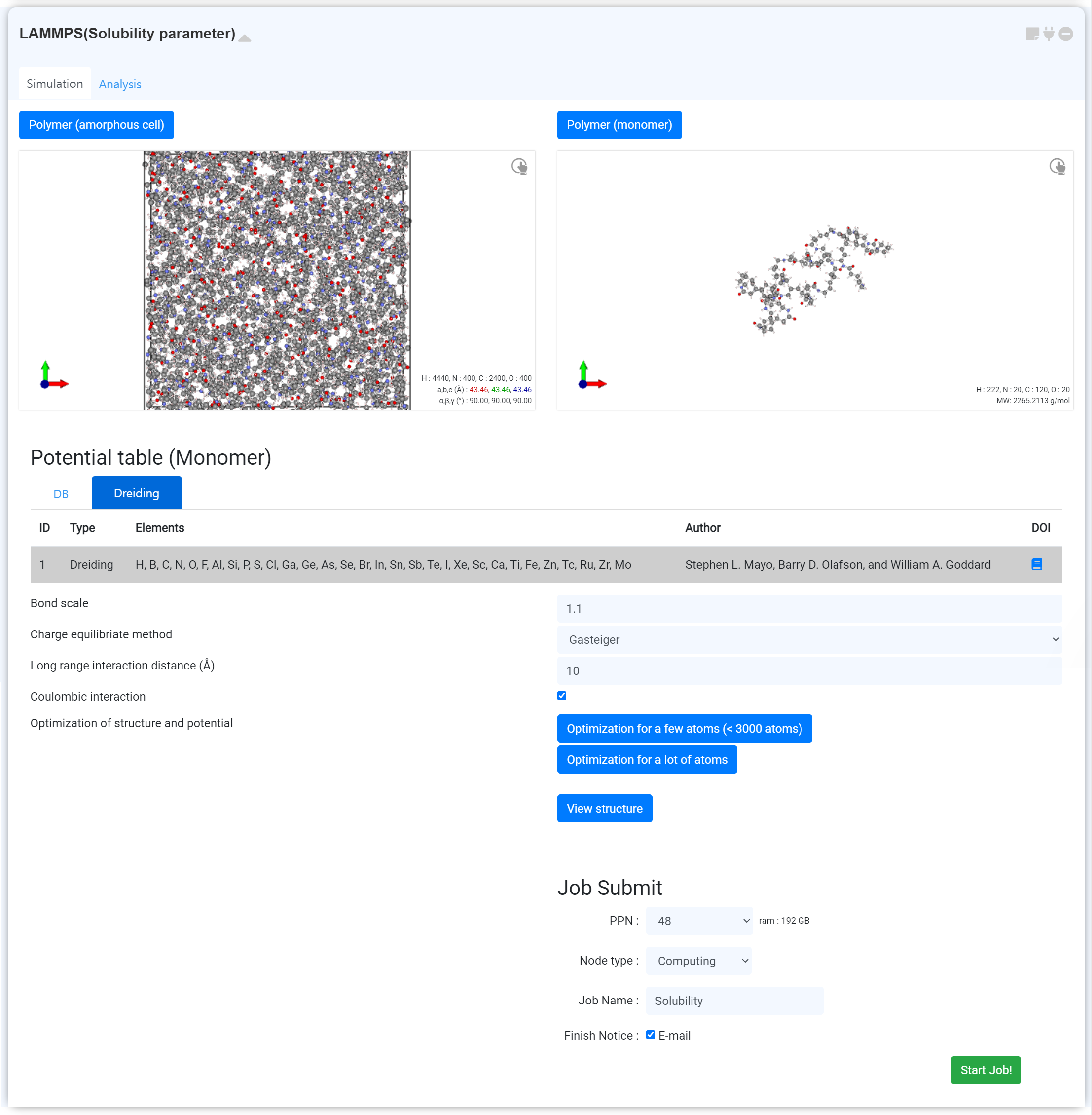
Solubility 계산을 위해, Amorphous cell과 monomer 구조가 필요합니다. 구조는 연결을 통해 불러올 수 있습니다. 연결 가능한 모듈 목록은 다음과 같습니다.
Amorphous cell은 liquid 영역의 polymer cell을 사용하는 것이 좋습니다.
이후 작업 이름을 설정하고 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
Visualizer에 구조 파일을 직접 업로드하면 계산이 정상적으로 진행되지 않습니다.
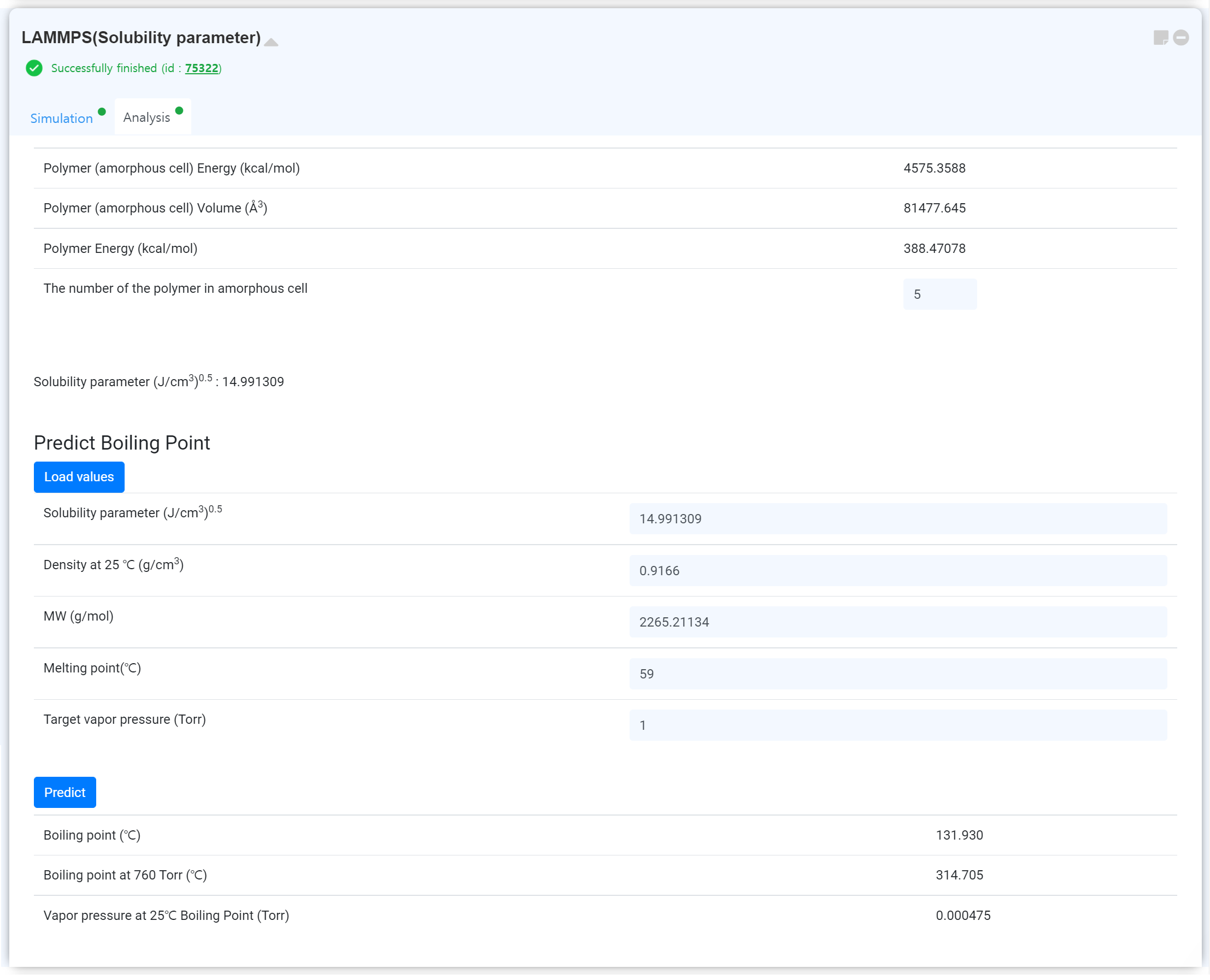
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
'The number of the polymer in amorphous cell' 의 값을 수정하며 달라지는 Solubility parameter 값을 확인할 수도 있습니다.
기계학습으로 끓는점을 예측할 수 있습니다. 를 눌러 계산값을 불러오고, 를 눌러 예측을 수행하십시오.
Solubility 계산을 위해, Amorphous cell과 monomer 구조가 필요합니다. 구조는 연결을 통해 불러올 수 있습니다. 연결 가능한 모듈 목록은 다음과 같습니다.
Amorphous cell은 liquid 영역의 polymer cell을 사용하는 것이 좋습니다.
- LAMMPS (Thermalization)
- LAMMPS (Tg/CTE)
- Structure Builder
- Molecule Builder
이후 작업 이름을 설정하고 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
Visualizer에 구조 파일을 직접 업로드하면 계산이 정상적으로 진행되지 않습니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
'The number of the polymer in amorphous cell' 의 값을 수정하며 달라지는 Solubility parameter 값을 확인할 수도 있습니다.
기계학습으로 끓는점을 예측할 수 있습니다. 를 눌러 계산값을 불러오고, 를 눌러 예측을 수행하십시오.
평형 분자동역학 (equilibrium molecular dynamics simulation) 계산을 통해 고분자 구조의 점도를 계산합니다. 이 모듈을 통해 재료가 갖는 평균 점도를 계산할 수 있습니다.
LAMMPS (Viscosity EMD) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
각 온도별로 시간에 따른 점도 데이터를 볼 수 있습니다.
LAMMPS (Viscosity EMD) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
- 계산의 정확도를 설정하십시오. 이는 데이터의 개수에 영향을 미칩니다. 다시 말해, 데이터 사이의 온도 간격을 결정합니다.
- 시뮬레이션을 수행할 시작 온도 및 종료 온도를 결정하십시오.
- 필요한 경우, Advanced Options를 열어 계산 조건을 보다 상세히 설정하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
각 온도별로 시간에 따른 점도 데이터를 볼 수 있습니다.
Strain rate를 조절 가능한 비평형 분자동역학 (non-equilibrium molecular dynamics simulation) 계산을 통해 고분자 구조의 점도를 계산합니다.
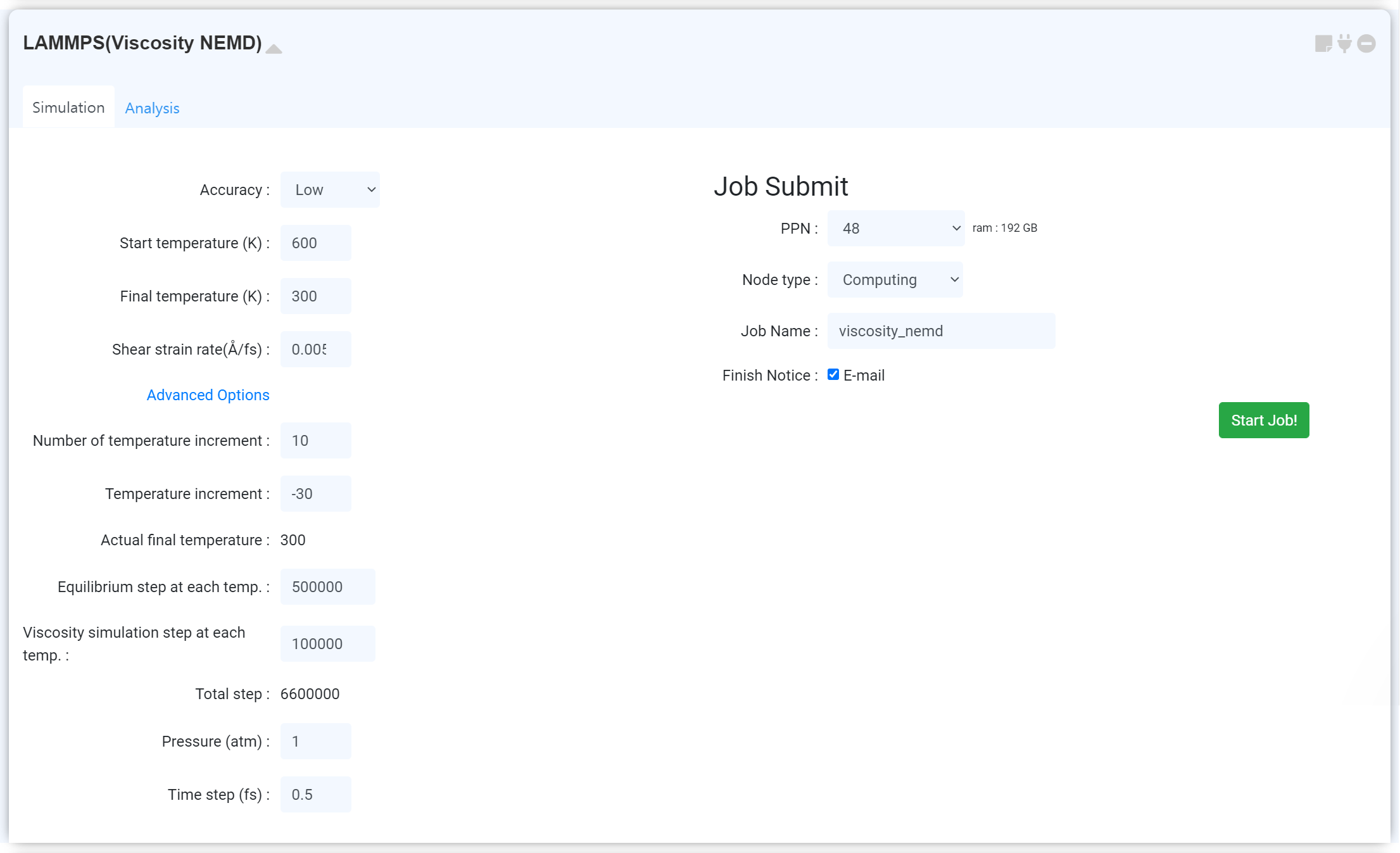
LAMMPS (Viscosity NEMD) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
각 온도별로 시간에 따른 점도 데이터를 볼 수 있습니다.
LAMMPS (Viscosity NEMD) 모듈은 Thermalization에 이어 수행하는 restart 모듈입니다. 모듈 추가 후, 계산이 종료된 LAMMPS (Thermalization) 에 연결하십시오.
Forcefield 등의 정보는 연결된 Thermalization 모듈로부터 가져와집니다.
- 계산의 정확도를 설정하십시오. 이는 데이터의 개수에 영향을 미칩니다. 다시 말해, 데이터 사이의 온도 간격을 결정합니다.
- 시뮬레이션을 수행할 시작 온도 및 종료 온도를 결정하십시오.
- 필요한 경우, Advanced Options를 열어 계산 조건을 보다 상세히 설정하십시오.
- 작업 이름을 설정하십시오.
- 'Start Job!' 버튼을 눌러 계산을 시작하십시오.
계산이 종료된 후, Update 버튼을 눌러 모듈을 업데이트하십시오. Analysis 탭에서 결과를 확인할 수 있습니다.
각 온도별로 시간에 따른 점도 데이터를 볼 수 있습니다.
Calphad 모듈은 상태도 (phase diagram)를 그리기 위한 모듈입니다. 보다 자세한 설명 및 데이터베이스 정보, 예제 계산은
appendix에서 확인하실 수 있습니다.
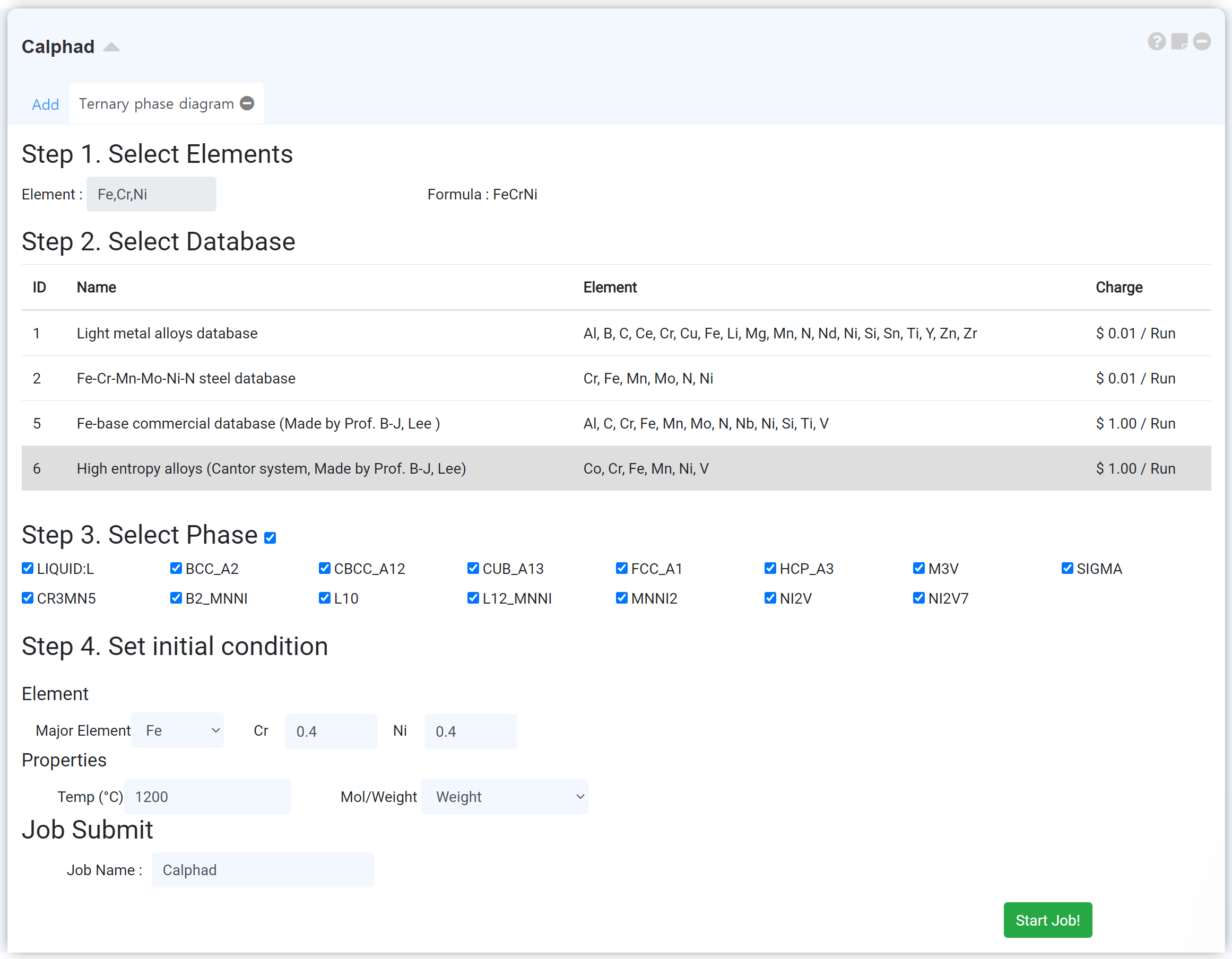
-
Phase diagram을 그리고자 하는 원소를 선택하십시오. Binary, ternary phase diagram의 경우 원소가 충분히 선택되면 주기율표가 자동으로 닫힙니다. -
해당 시스템에 가장 적절한 데이터베이스를 선택하십시오. 데이터베이스에 대한 자세한 정보는 appendix를 참고하십시오. -
Phase diagram을 그리기 원하는 상을 선택하십시오. -
Phase diagram이 그려질 초기 시작 지점을 지정하십시오. 적절한 시작 지점이 아닐 경우 제대로 된 그래프가 그려지지 않을 수 있습니다.
재료과학 분야의 대표적인 예측 모델인 Crystal Graph Convolutional Neural Network (CGCNN)은 재료의 구조를 입력값으로 활용하여 재료의 물성을 결과값으로 예측하는 정방향 (direct) 예측 모델입니다. CGCNN 모듈은 CGCNN 모델을 사용하여 기계학습 계산을 수행할 수 있게 만든 모듈입니다. 이 모듈을 사용하면 사용자는 원하는 재료의 물성 (formation energy, band gap, bulk/shear moduli, poisson ratio 등)을 예측할 수 있습니다.
트레이닝 데이터셋을 업로드하고 확인할 수 있는 탭입니다. Upload zip file 버튼을 클릭하여 데이터베이스 압축 파일을 업로드하십시오. 데이터셋은 다음 예시를 참고하여 구성할 수 있습니다. 실제 학습 시에는 더 많은 수의 샘플이 필요합니다.
나만의 기계학습 모델을 만들 수 있는 영역입니다. 학습 데이터와 테스트 데이터를 어떻게 분배하여 기계 학습 모델을 만들 것인지, 모델을 만드는 데 필요한 하이퍼 파라미터를 어떻게 조절할 것인지, 그리고 학습에 어떠한 최적화 방법을 사용할 것인지 등을 결정하여 나만의 기계학습 모델을 만들 수 있습니다.
Dataset ratio 에서는 내가 가지고 있는 데이터 개수를 어떻게 나눠 활용할 것인지를 결정합니다. 만일 위 그림과 같이 설정할 경우, 데이터 중 약 60%는 Training-set에서 x,y의 상관관계를 찾는 학습에 사용하고, 나머지 20%를 Validation에 사용해 찾아낸 관계가 맞는지 확인합니다. 나머지 20%는 실전에 쓸 수 있도록 Test-set에 적용합니다.
이외에도 네트워크와 관련된 Layers, optimize와 관련된 Other options가 있습니다.
모든 설정을 완료했다면 작업 제출 (Job submit) 문서를 참고하여 작업을 제출하십시오.
계산이 종료된 후, CGCNN로 얻은 학습 결과를 보여주는 탭입니다. 내가 설계한 기계학습 모델이 학습/테스트 데이터를 얼마나 정확히 예측하는지 학습 단계에 따른 Epoch를 확인할 수 있습니다. 일반적으로는 학습 데이터의 정확도가 테스트 데이터의 정확도보다 높기 때문에, 습득한 결과에서 학습 데이터의 정확도가 너무 높다면 과적합 (overfitting) 을 해결할 방법을 찾아야 합니다.
학습 종료 후, 계산된 모듈을 불러와서 업로드된 다른 구조의 물성을 예측할 수 있는 탭입니다. Upload zip file 버튼을 클릭하여 데이터베이스 압축 파일을 업로드하십시오. 데이터셋은 다음 예시를 참고하여 구성할 수 있습니다.